Genomic insights into multidrug-resistant Acinetobacter baumannii strains isolated from Dhaka, Bangladesh
Abstract
Acinetobacter baumannii is one of the most common nosocomial infections causing pathogens. Multidrug-resistant (MDR) A. baumannii is a growing concern to the healthcare system in Bangladesh, necessitating the elucidation of the genetic characteristics of this species. Five MDR, biofilm-forming A. baumannii strains isolated from clinical samples in Dhaka, Bangladesh, underwent a comprehensive whole-genome shotgun sequencing. In silico analyses determined strain types, antibiotic resistance, and virulence genes. The phylogenomic and pan-genome comparative analysis included the study samples along with an additional 15 Bangladeshi A. baumannii strains collected from the public database. The antibiotic-resistant profile showed that all the strains were resistant to meropenem. Two strains were resistant to polymyxin B up to 64 µg/ml. The average draft genome size of the strains was 3.93 Mbp, with about 39% GC content. Multilocus sequence typing characterized the strains as ST1, ST2, ST164, and ST623. All strains carried different classes of β-lactamase genes, including the carbapenem hydrolyzing enzyme coding genes blaoxa-23 and blaoxa-51. One of the five strains studied harbored blaNDM-9 and blaNDM-23 genes. The phosphotransferase group of aminoglycosides modifying enzymes was the most common. All the strains had relatively similar virulence profiles. Pan-genome analysis revealed 1944 core and 1302 unique genes. Phylogenomic analysis revealed the distribution of these strains in different clusters with other MDR strains isolated worldwide. The study results suggest the implementation of antibiotic stewardship and genomic surveillance of MDR A. baumannii to control the spread of antibiotic resistance in Bangladesh.
INTRODUCTION
Antimicrobial resistance (AMR) presents a serious global health challenge, responsible for millions of deaths annually [1]. The widespread and often unregulated use of antibiotics in humans, farm animals, and agriculture increases AMR risks, affecting both human and animal health. Although considerable progress has been made in developing antimicrobial agents over recent decades, the rapid rise of multidrug-resistant (MDR) bacteria continues to complicate infectious disease management worldwide [2,3]. Given the rising prevalence of MDR pathogens, there is an urgent need to address this problem by resistant bacteria like A. baumannii across healthcare, agriculture, and environmental sectors [4].
A. baumannii is a Gram-negative opportunistic human pathogen responsible for nosocomial infection in hospital settings and intensive care units with a significant impact on mortality rates [5]. Several other studies have confirmed that A. baumannii, along with other clinically relevant Gram-negative bacteria, exhibited a marked decline in sensitivity to most antibiotics over the past two decades [6,7]. Clinical isolates of Acinetobacter showed resistance to gentamicin (92.7%) and ceftriaxone (87.8%), imipenem (56%), and even colistin (19.5%) which is considered the last resort of treatment due to its high toxicity [4]. It was reported that more than half of these pathogens produced ß-lactamases and carbapenemases, resulting in resistance to the ß-lactam and carbapenem group of antibiotics [8].
A. baumannii is one of the most common pathogens causing nosocomial infections in Bangladesh and has recently emerged as an extensive drug-resistant in many clinical cases [4,8–10]. According to a study [9], the three most common pathogens isolated from patients with nosocomial infections in Bangladesh are Klebsiella pneumoniae, A. baumannii, and Escherichia coli. Although the antibiotic resistance pattern is established, little is known about the genomic makeup of this pathogen found in Bangladesh. The difficulty of treating resistant A. baumannii has made it urgent to understand and analyze the whole genomic sequence of this bacterium to treat and prevent the spread of hospital-acquired infections effectively. Hence, the primary objective of our research is to ascertain the genomic composition of A. baumannii strains obtained from healthcare facilities located in Dhaka, Bangladesh, to gain a more comprehensive understanding of the genetic variability and adaptability of A. baumannii.
MATERIALS AND METHODS
Sample collection and identification
A total of five clinical isolates (SDL03, SDL07, SDL32, SDL35, and SDL40) of A. baumannii were collected from Dhaka, Bangladesh (Table 1). The samples were collected in T1N1 soft agar (Oxoid, UK) from the hospital facilities and transferred to the Department of Biochemistry & Microbiology, North South University, where they were grown in MacConkey agar (Oxoid, UK). The molecular confirmation of the pure colony of strains was done by a conventional 16s rRNA gene sequencing using universal primer set 27F/1492R [11]. The amplified 16s rRNA gene sequences were analyzed using the Basic Local Alignment Search Tool (BLAST) to confirm the identification of the obtained samples as A. baumannii. The sample acquisition and subsequent use were approved by the Institutional Review Board, with approval number 2019/OR-NSU/IRB-No.0504.
Antibiotic susceptibility test
The antibiotic resistance patterns of the confirmed A. baumannii strains were determined by the Kirby-Bauer disc diffusion method, as stated in the Clinical and Laboratory Standards Institute (CLSI) guidelines [12]. In this study, antibiotics from various classes were chosen based on the current national guidelines for treating A. baumannii infections [13]. This selection aimed to assess bacterial resistance profiles and establish whether the isolates met the criteria for MDR or extensive drug resistance (XDR) [14]. The antibiotic disks used in this study were amikacin (AMK 30 µg), gentamicin (GEN 10 µg), imipenem (IPM 10 µg), meropenem (MEM 10 µg), ceftriaxone (CRO 30 µg), cefepime (FEP 30 µg), ciprofloxacin (CIP 5 µg), levofloxacin (LVX 5 µg), trimethoprim-sulfamethoxazole (SXT 25 µg), ampicillin (AMP 10 µg), piperacillin-tazobactam (TZP 110 µg), and tigecycline (TGC 15 µg) (Oxoid, UK). Additionally, the minimum inhibitory concentration (MIC) against colistin and polymyxin-B (Sigma-Aldrich, USA) of all A. baumannii strains were determined by broth-microdilution (BMD) quantitatively, following the CLSI guidelines [12]. All experiments were performed in triplicate on three separate occasions. Here, E. coli DH5α, a strain sensitive to colistin, was used as a control strain [15] to ensure test accuracy. Cation-adjusted Mueller-Hinton broth (Oxoid, UK) acted as a negative control to ascertain suitable conditions for reliable MIC determination.
Biofilm formation assay
The biofilm production ability of the studied strains was determined by the qualitative and quantitative methods described with slight modifications [16]. The biofilm formation was determined by using the following methods: ODcut = ODavg of negative control + 3 × standard deviation (SD) of OD of negative control; OD ≤ ODcut = non-biofilm former; ODcut < OD ≤ 2 × ODcut = weak-biofilm former; 2× ODcut < OD ≤ 4 × ODcut = moderate-biofilm former; OD > 4 × ODcut = strong-biofilm former [17]. Briefly, a single colony from each culture plate of the samples was inoculated and incubated overnight at 37°C in a shaking incubator at 220 rpm. A 1:1000 dilution of the inoculum in the nutrient broth was incubated again at 37°C for 48 hours in a static place. The presence of biofilm was determined by 0.1% crystal violet staining. The biomass was solubilized using 30% acetic acid to quantify the biofilm. The absorbance of the solubilized biomass was measured at 590 nm using a microplate reader (Multiskan EX, Thermo Scientific, Finland) (Supplementary Table 1). All the processes were carried out at room temperature and were repeated four times under the same conditions. Furthermore, a bivariate analysis was conducted to examine the correlation between the degree of biofilm formation and antibiotic resistance patterns across all five studied strains (Supplementary Table 2), using SPSS Version 26.0 (IBM Corp., NY, USA) and following the protocol outlined by Ullah et al. [18].
DNA extraction and preparation for whole genome sequencing
Bacterial genomic DNA was extracted using Wizard® Genomic DNA Purification Kit according to the manufacturer’s instructions (Promega, USA). The quantity and quality of extracted DNA was determined using a NanoDrop™ 2000 (Thermo Scientific, USA).
Sequence assembly and annotation
The A. baumannii clinical strains were subjected to Whole Genome Shotgun (WGS) sequencing in an Ion GeneStudio™ S5 System (Thermo Fisher Scientific, USA) by DNA Solution Ltd., Dhaka, Bangladesh. Quality control and adapter trimming were performed for the multiple reads generated, as described [19]. Multiple reads were generated for each sample, with quality control and adapter trimming carried out using Torrent Suite™ Software version 5.10.0. The raw reads were uploaded to the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) [20] server and assembled using Unicylcer v0.4.8 [21], Spades v3.13.0 [22], Bandage v0.8.1 [23], Quast v5.0.2 [24], and Samtools v1.3_31 [25]. The assembled genome sequences were uploaded to the National Center for Biotechnology Information (NCBI) genome sequence database under the BioProject number PRJNA823880 and further annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [26]. Proksee, a comprehensive system utilized for assembly, annotation, analysis, and visualization purposes, was employed to create a circular map representing the local alignment of the strains [27] (Supplementary Figure 1).
Predictions of antibiotic resistance genes
Antibiotic resistance genes (ARGs) were predicted from each annotated nucleotide sequence based on homology and single nucleotide polymorphism (SNP) models by using the Resistance Gene Identifier (RGI) tool of the Comprehensive Antibiotic Resistance Database (CARD) with the default parameters [28], except for including nudge ≥95% identity loose hits to strict. A heatmap was generated from http://www.bioinformatics.com.cn/srplot, an online platform for data analysis and visualization, using the predicted ARGs of the five A. baumannii strains used in this study and publicly available WGS data of 15 other A. baumannii strains isolated from Bangladesh (Supplementary Table 3) [29,30].
Genomic characterization
Multilocus Sequence Typing (MLST) 2.0.9 tool in the Center for Genomic Epidemiology Server [31,32] was employed with the Pasteur MLST scheme for A. baumannii to determine the sequence type (ST) of each sequence. The insertion sequences (IS) and prophages in bacterial genomes offer critical insights into the mechanisms behind bacterial evolution, adaptation, and AMR - key factors in controlling bacterial infections and developing effective antimicrobial strategies. The presence of any IS elements and prophage regions were predicted using ISFinder [33] and PHASTER [34], respectively. Both the tools predicted the presence and position of IS elements or prophages in the corresponding contigs of the assembled sequences. The pathogenicity was determined using PathogenFinder 1.1, also from the Center for Genomic Epidemiology [35]. The presence of pathogenicity islands was analyzed using IslandViewer 4 [36]. Pan-core genome analysis was performed to understand the genetic variation and evolutionary insights of the five strains included in this study plus 15 publicly available A. baumannii genome sequences in the NCBI genome database from Bangladesh using the Bacterial Pan-Genome Analysis tool (BPGA), version 1.3 [37].
Phylogenomic tree construction
The Average Nucleotide Identity (ANI) of the sequences of this study was calculated using FastANI v1.3 [38] (Supplementary Figure 2). From the ANI match, complete sequences were selected with a cutoff score of >97%. The genome BLAST Distance Phylogeny (GBDP) strategy was used by the Type strain genome server (TYGS) to contract the phylogenomic tree using 50 strains of A. baumannii (Supplementary Table 3) [39] utilizing default parameters followed by annotating the tree in Interactive Tree of Life (iTOL) [40].
RESULTS
MDR phenotype and biofilm production rate among A. baumannii strains
The antibiotic susceptibility test revealed high resistance against all the major antibiotic groups except polymyxins and tetracycline. Tigecycline and polymyxin B appeared to be the most effective antibiotics against studied A. baumannii isolates (Figure 1). However, two of these isolates (SDL32 and SDL40) exhibited resistance to polymyxin B. The disk diffusion test results for five studied A. baumannii strains against various antibiotics are shown in Supplementary Figure 3. As all the isolates in this study were resistant to at least three classes of antibiotics, they are characterized as MDR [14].
Besides, susceptibility towards the polymyxin (polymyxin B, colistin) group of antibiotics was determined by the BMD method (Figure 1). The MIC values for colistin against strains SDL03, SDL07, and SDL32 were 0.5 mg/L, 1 mg/L, and 0.5 mg/L, respectively, all were below 2 mg/L. In contrast, strains SDL35 and SDL40 had MIC values of 2 mg/L. According to earlier CLSI guidelines, MIC values <2 mg/L were categorized as colistin-susceptible. However, CLSI (2020) revised this classification, removing the ‘susceptible’ category for Acinetobacter spp. and defining MIC ≤2 mg/L as intermediate and MIC ≥4 mg/L as resistant. For Polymyxin B, strains SDL03, SDL07, and SDL35 were classified as sensitive, with an MIC of 2 mg/L. In contrast, strain SDL32 (MIC = 32 mg/L) and strain SDL40 (MIC = 64 mg/L) were resistant to Polymyxin B. Later, the biofilm formation ability of the samples was determined by crystal violet staining (Table 1 & Supplementary Table 1). It was observed that strains would form biofilm regardless of their biological sources; however, the A. baumannii isolates collected from the respiratory tract area were found to form more biomass than the other two samples collected from the urine or wound swab.
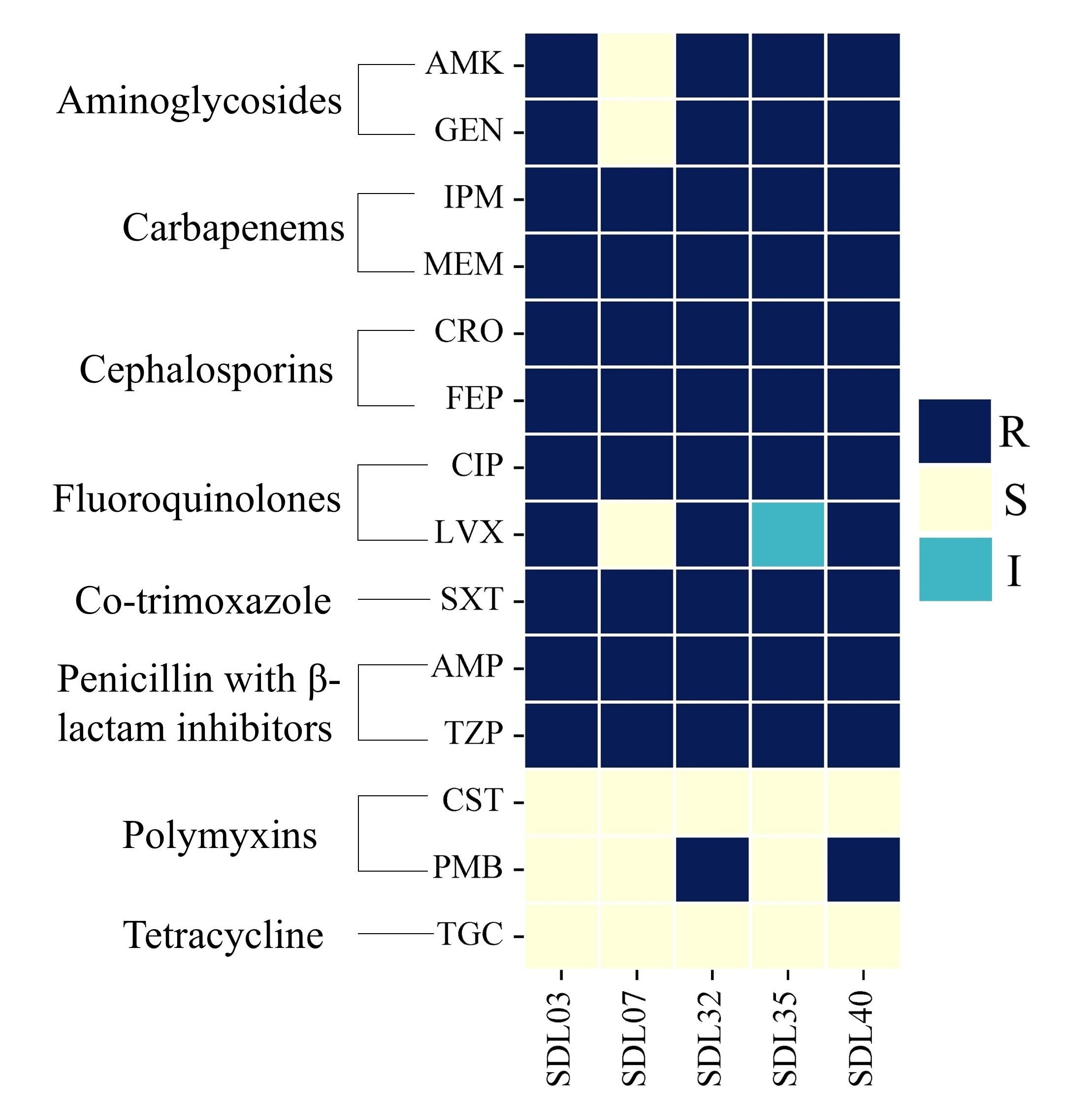
Table 1. Biofilm formation ability of A. baumannii.
General features of the sequenced strains
In the WGS, the raw sequences were generated with coverage higher than the threshold 20X, with an average GC content of approximately 39%. The quality of the assembled genomes was assessed using A. baumannii K09-14 as a reference strain. Once assembled and annotated, the sequences were uploaded to the NCBI database to generate unique accession numbers for all the samples. The general features of the sequencing data are presented in Table 2 and Supplementary Figure 1.
Table 2. General characteristics of the sequence output of the studied strains and reference strain A. baumannii K09-14.
Prediction of antibiotic resistance genes
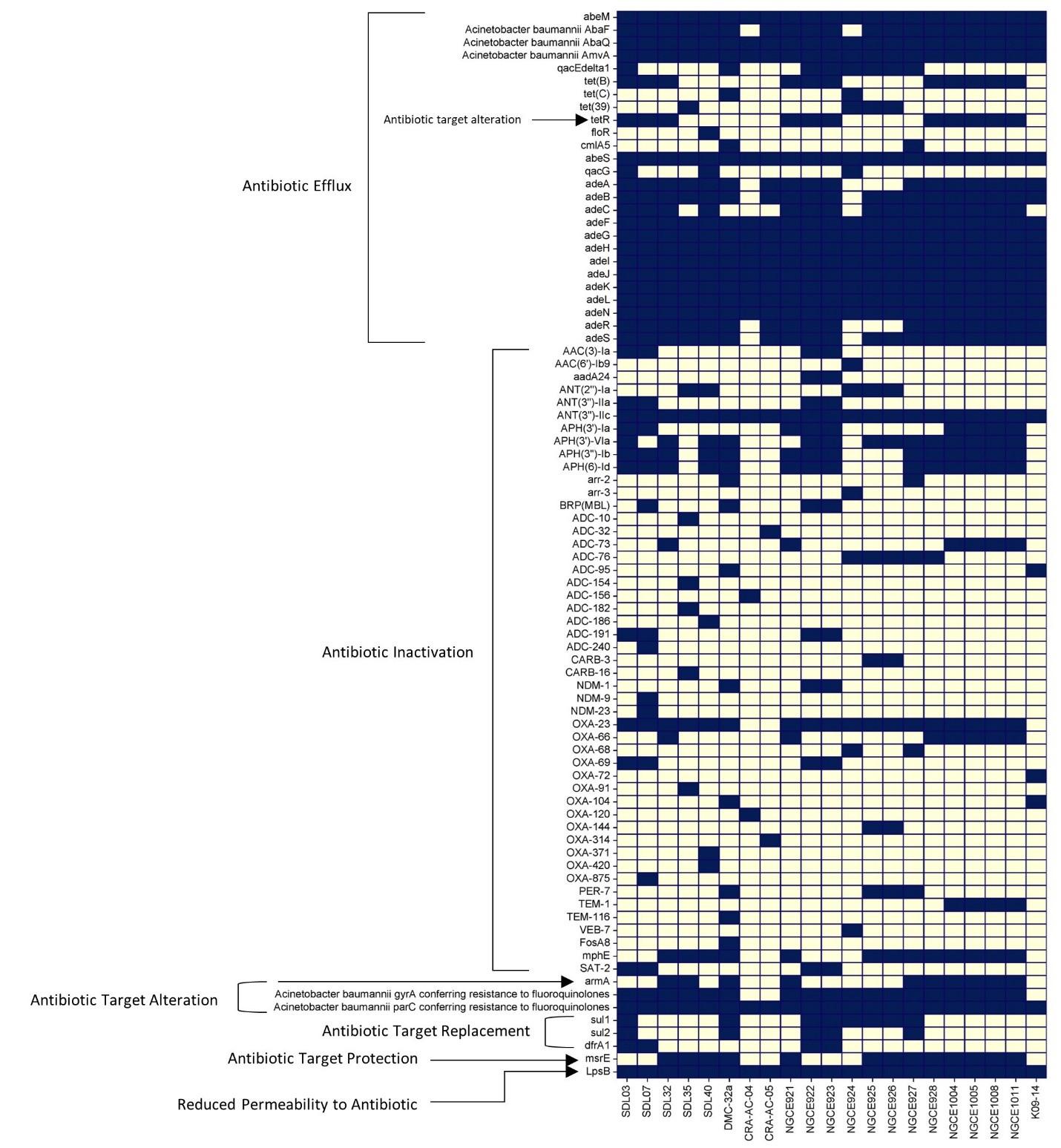
The annotation of the genome sequences revealed the presence of many well-characterized ARGs, shown in Figure 2. All the Bangladeshi strains contain different ARGs from all the ARG groups, and these genes are widely distributed among the strains. There is no significant difference in ARG number and/or distribution among the Bangladeshi strains, except that the genes responsible for the inactivation of antibiotics are present unevenly among these strains. Efflux genes seem to be more predominant than other classes of genes across the strains. All the Bangladeshi strains except two harbor the blaoxa-23 gene, whereas strain SDL07 harbored blaNDM-9 and blaNDM-23 genes. Other β-lactamase classes of ARGs are present infrequently in the phenotypically carbapenem-resistant strains.
Presence of different classes of virulence factor genes
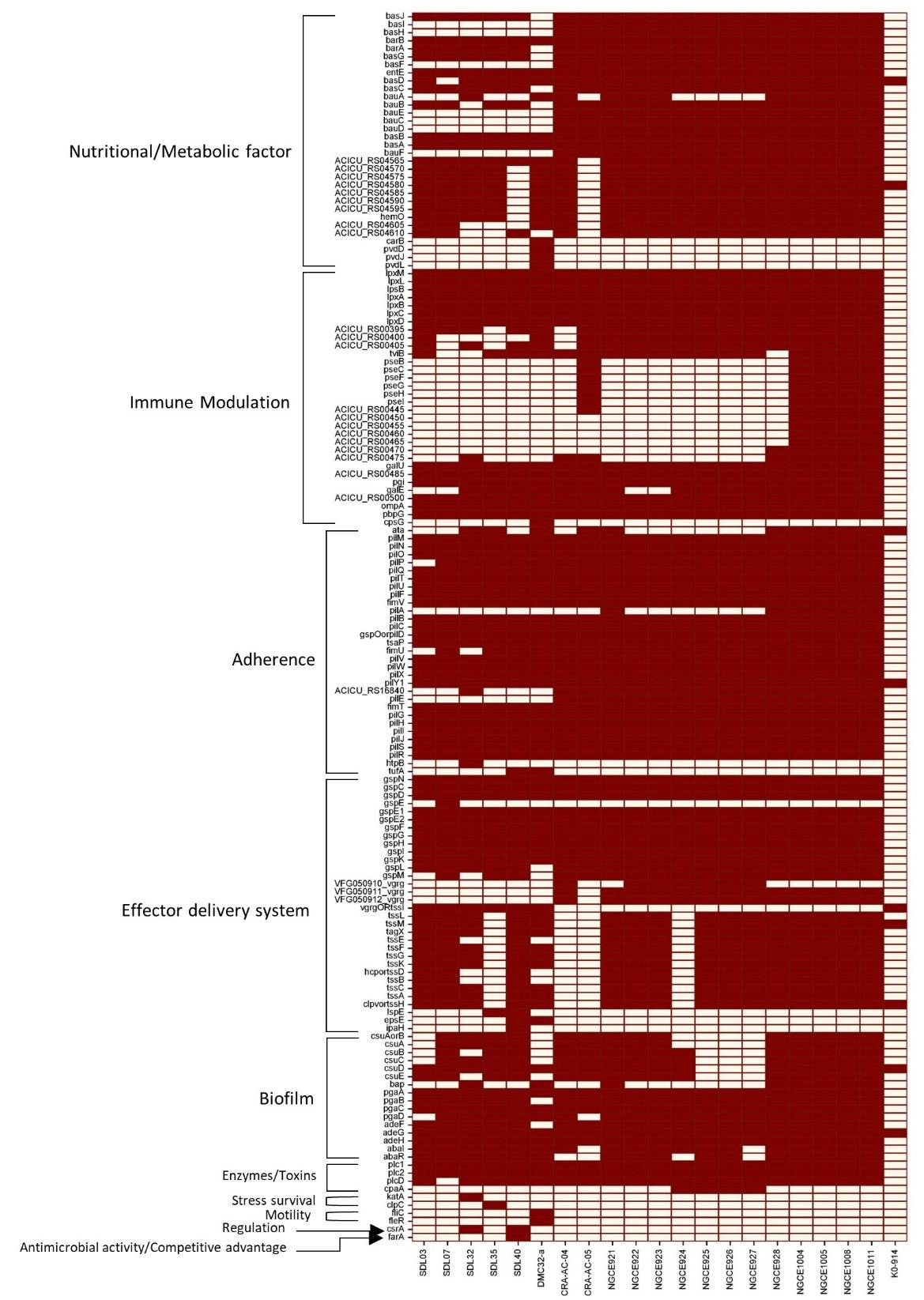
The presence of virulence factors was also analyzed, and it was found that all the Bangladeshi strains contain different kinds of virulence factors compared to the reference strain (Figure 3). While virulence factors significant for adherence, effector delivery system, and biofilm formation were present in all strains, comparatively fewer immune modulation factors were present in the studied strains. Again, a sporadic pattern can be seen in the presence of the virulence factors responsible for a major iron uptake regulating siderophore and acinetobactin formation among the five strains of this study. Furthermore, most of the studied strains harbored a higher number of pathogenicity islands compared to the reference strain K09-14 (Table 2).
Phylogenomic tree analysis
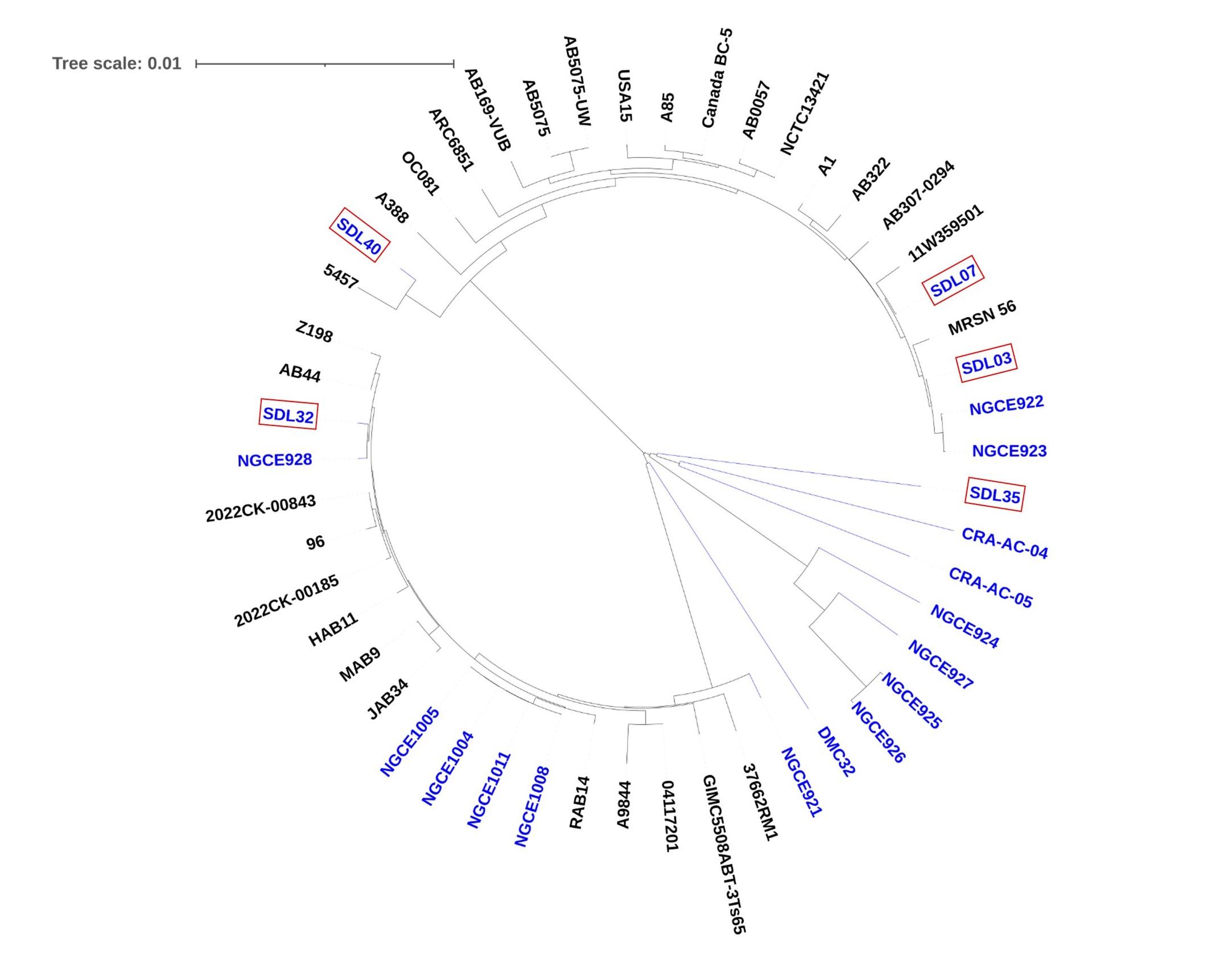
A phylogenomic analysis was conducted to identify the evolutionary relationship among the strains isolated from Bangladesh and other MDR A. baumannii isolated worldwide, as shown in Figure 4. The phylogenomic tree demonstrates that Bangladeshi Acinetobacter strains are genetically diverse and distributed across different evolutionary branches. Such dispersion of these strains among non-Bangladeshi isolates implies that these Acinetobacter strains from Bangladesh are from multiple evolutionary origins, suggesting that they may have been introduced from different sources or evolved separately within distinct lineages. This finding also aligns with the concept that these strains may have acquired MDR independently or adapted to similar environmental pressures that drive their divergence.
Pan-core genome analysis
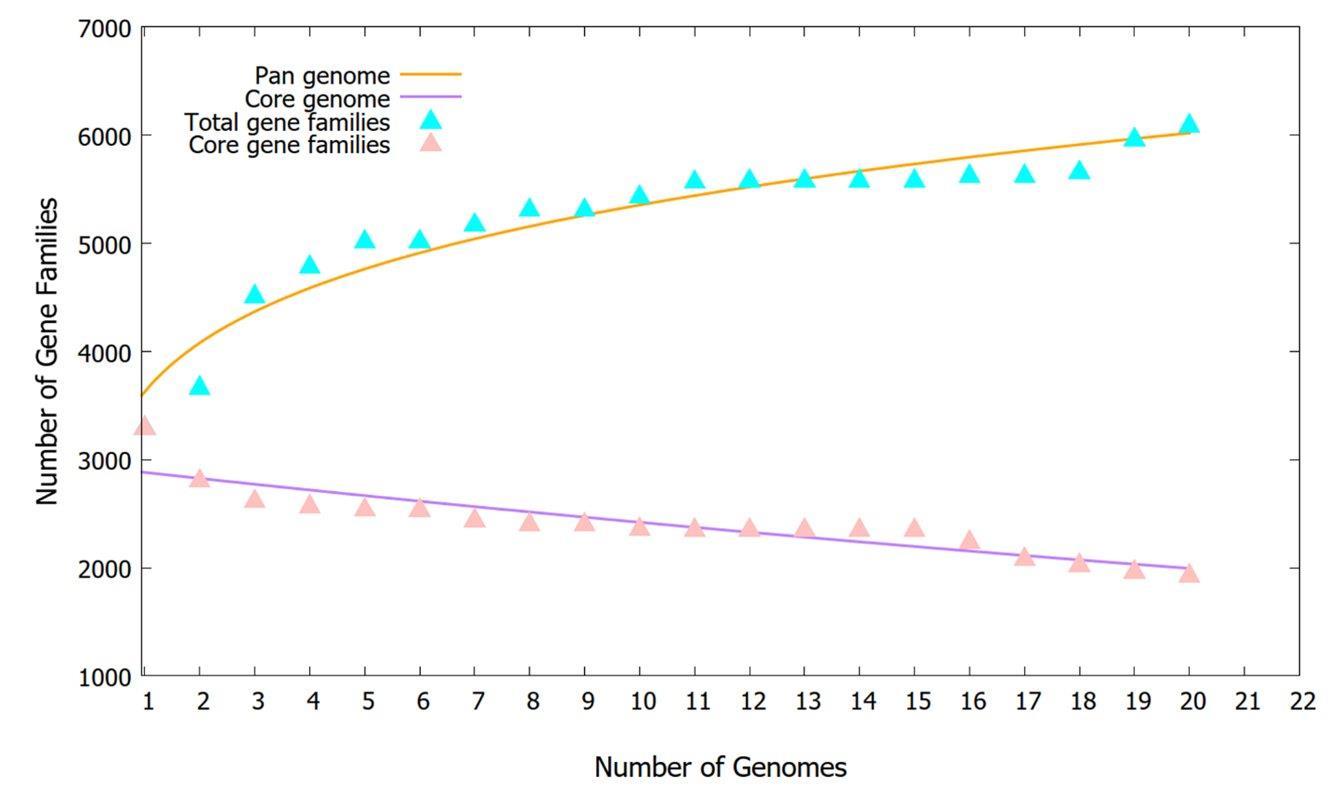
A pan-genome analysis of twenty A. baumannii strains was conducted, which comprised fifteen recently sequenced A. baumannii genomes originating from Bangladesh alongside our five studied strains. The pan-core plot (Figure 5) shows an increase in the total gene numbers, indicating the emergence of new genes by introducing a new genome to the pan-genome analysis. A total of 1944 genes were detected as core genes, shared by all the strains. There were 510 exclusively absent genes in the genome of A. baumannii; additionally, there were 1434 ± 182 accessory genes in the genome (Supplementary Table 4). Moreover, 1302 unique genes were detected in the pan-genome, where DMC32 was found to have acquired the highest number of unique genes (307), followed by SDL35 (296), whereas few strains lacked any unique genes. This observation is further supported by the fact that 82% of the unique genes identified in five specific A. baumannii strains: CRA-AC-04 (n=181), CRA-AC-05 (n=159), DMC32 (n=307), SDL35 (n=296), and SDL40 (n=128). In contrast, strains NGCE922, NGCE923, NGCE926, NGCE1004, NGCE1005, NGCE1008, and SDL07 did not harbor any unique genes. The functional adaptations of A. baumannii showed Bpan values of 0.183883 (i.e., <1) in the power-law regression model, suggesting that the pan-genome is open but may be closed soon [41].
DISCUSSION
A. baumannii strains pose a significant threat in Bangladesh, often exhibiting MDR or XDR traits [4,7,42]. Despite this, the genomic characteristics of Bangladeshi A. baumannii strains remained largely unexplored until recently [29], when they focused only on the distribution of A. baumannii strains in different clonal complexes. However, this gap limited our understanding of the antibiotic resistance mechanisms and virulence among Bangladeshi A. baumannii strains. We have sequenced the whole genomes of five clinical A. baumannii isolates and compared them to 15 additional A. baumannii strains from Dhaka to address this gap. WGS analysis showed that our five MDR strains harbored diverse resistance genes and virulence factors. The strains SDL03 and SDL07 belong to the clonal population of ST1 according to the Pasteur scheme of MLST typing (Table 2). SDL32 was found to be in ST2. The strains isolated from Bangladesh in different studies also belonged to ST1 and ST2 [10,29]. The remaining sequence types, ST164 and ST623 in strains SDL35 and SDL40, respectively, were reported globally but uncommon in Bangladesh [43,44]. Overall, this study sheds light on the unique genomic makeup of MDR A. baumannii strains in Bangladesh, highlighting their potential for diverse antibiotic resistance and virulence and paving the way for further investigation into their epidemiology and control strategies.
The studied strains are MDR in nature, exhibiting resistance to different classes of conventionally used antibiotics like β-lactams, carbapenems, aminoglycosides, and fluoroquinolones (Figure 1). Second-line antibiotics like polymyxin and tigecycline were effective against most of our isolates; however, two strains, SDL32 and SDL40, displayed resistance to polymyxin B. Only one strain, SDL07, was identified as susceptible to a third-generation fluoroquinolone, levofloxacin. This, coupled with non-susceptibility towards last-resort antibiotics, shows the emergence of XDR and further emphasizes the importance of understanding the molecular mechanism of drug resistance by analyzing the whole genome.
Diverse resistance genes were identified using the CARD database, which can render resistance through antibiotic inactivation, target protection/modification, and efflux pumps. The phenotypic resistance profile observed (carbapenems, cephalosporins, fluoroquinolones, aminoglycosides) is on par with the genotypic findings illustrated in Figure 2. A common thread emerged by comparing the data with a broader set of Bangladeshi strains: each isolate harbored at least one gene from each group of ARG families. The presence of Aminoglycoside Nucleotidyltransferase, ANT (3”) enzymes associated with resistance to streptomycin and spectinomycin [45], are present in all the strains used in the comparative analysis. Previous studies reported the frequent presence of genes like aac(3)-I, aac(6′)-Ib, aph(3′)-I, aph(3′)-IIb, aph(3′)-VIa, aph6, and ant(3”)-I in aminoglycoside-resistant strains found in Asia, often showcasing multiple coexisting genes associated with higher resistance levels [46,47]. Our analysis revealed a similar pattern, with diverse Aminoglycoside Phosphotransferases APH groups detected in our isolates and the other Bangladeshi strains (Figure 2). However, the aminoglycoside acetyltransferase AAC group of genes was less prevalent in Bangladeshi strains despite these strains being resistant to both amikacin and gentamicin. These findings are suggestive of the differences in resistance mechanisms prevalent among the strains of this region.
Another set of antibiotic-inactivating enzymes encoded by the different classes of β-lactamase can directly hydrolyze carbapenem antibiotics and are present in the studied five strains. OXA-23-like β-lactamase (blaoxa-23 ) and OXA-51-like β-lactamase (blaoxa-66, blaoxa-69, blaoxa-91, blaoxa-371, and blaoxa-875) are present in all five strains, aligned with reports linking them to increased carbapenem resistance in A. baumannii [48]. The presence of B, C, and D β-lactamase classes like blaNDM, blaADC, and blaoxa-58, especially NDM-9 and NDM-23, was predicted using the CARD database. The enzymes that break down carbapenems (β-lactamase genes) are one of the causes of resistance, and other factors, such as efflux pumps and porins, make the problem even worse [49]. These groups of carbapenem resistance contributing genes are present in the studied strains (Figure 2), accounting for the elevated resistance against both imipenem and meropenem.
Acquired resistance in bacteria can also be caused by polymorphisms in antibiotic target genes, resulting in target alteration. In this study, all five strains harbor gyrA and parC genes with mutations in the Quinolone-resistance determining region (QRDR) associated with fluoroquinolone resistance [50]. However, no known polymorphisms in the pmrCAB operon, responsible for mediating resistance towards the polymyxin group of antibiotics [51,52], were found in SDL32 and SDL40 (Figure 1).
Our strains displayed polymorphisms in the AdeS and AdeR regulatory proteins of the AdeABC efflux pump that belong to the resistance-nodulation-cell division family. Previously, AdeRS polymorphism has been linked to increased AdeABC expression in European clones [53], which are highly resistant to carbapenems, fluoroquinolones, and aminoglycosides [54,55]. In addition to the presence of different aminoglycosides modifying enzymes, β-lactamases, and mutations in the QRDR region, the polymorphic AdeRS system in the studied strains (Supplementary Table 5) can potentially be the reason for resistance towards both second and third-generation antibiotics of multiple classes.
Understanding the virulent potential of these isolates is equally crucial as deciphering the resistance mechanisms. The ability of these MDR A. baumannii strains to cause infections and potentially evade host defenses was assessed by looking into the specific virulence factors associated with these strains, as shown in Figure 3. Notably, three isolates from respiratory tracts displayed strong biofilm production, which is concerning due to their potential to contribute towards ventilator-associated pneumonia and ICU settings [56–58]. However, no correlation was found between the biofilm-producing ability of the studied strains and their AMR patterns, possibly due to a small number of strains (Supplementary Table 2).
While our five strains contained lesser virulence factors responsible for immune modulation and metabolic factors, adherence and biofilm formation factors were uniformly present. The presence of these genes shows the potential for increased pathogenicity in the future. Therefore, further investigation into their evolutionary origins and spread is crucial. Our phylogenetic analysis offered valuable insights into this aspect. The comparative analysis in Figure 4 illustrates that despite being small in sample number, our studied strains are not grouped in a single cluster. Instead, they are present along with global strains in three different clades. The diverse distribution of strains across different clades suggests a complex global origin, further evident from the presence of one Global Clone 1 Lineage 3 strain (SDL40), originally tracked back to Indian strains [59]. The Bangladeshi strains, on the other hand, were found to be very closely related to each other even when in different clades, probably due to the fact that all of these were collected from the hospitals of Dhaka city. However, this does not imply the lack of diversity among the Bangladeshi strains. For example, SDL07 harbors two β-lactamase genes, blaNDM-23 and blaNDM-9, which are usually associated with K. pneumoniae; gene transfer might have occurred between these two bacterial species [60,61]. The pangenome analysis in Figure 5 shows the pangenome is still open, suggestive of ongoing evolution and gene gains, pointing towards a possibility of unique evolutionary trajectories.
Previous studies [29,30] and whole genome analysis of our five strains revealed that β-lactamase genes represent a key contributor to resistance in carbapenem-resistant A. baumannii; however, antibiotic resistance in this pathogen is not mediated by a single factor. Instead, multiple factors synergistically increase the resistance toward different classes of antibiotics. These observations emphasize the complex interplay of various resistance genes in mediating antibiotic resistance among Bangladeshi A. baumannii strains. While the overall pattern aligns with the previous report [29], our findings suggest differences in specific gene prevalence (Figure 2) and potential polymorphisms (Supplementary Table 5) within Bangladeshi strains. Due to the small number of strains, the observations present merely association, not definite causation of resistance. However, this is one of the first genomic studies on A. baumannii in Bangladesh. Thus, it will provide comprehensive genomic information and insights into common factors in drug-resistant A. baumannii.
CONCLUSIONS
This study aimed to look deep into the genetic makeup of different elements of five MDR A. baumannii with the help of whole genome sequencing. Here, 5 Bangladeshi strains were compared with 15 other Bangladeshi A. baumannii strains. While a larger, more diverse strain collection could further enhance the depth and generalizability of findings, even limited sample sizes can reveal meaningful trends and foundational knowledge. It was revealed that the bacteria possess both ARGs and virulence factors. The phenotypical observation of high resistance could be explained by the numerous antibiotic-resistant genes of all groups present alongside several SNPs/polymorphic genes. The presence of biofilm as a virulent factor indicates this nosocomial pathogen’s dissemination in healthcare setup by persisting on different abiotic medical devices. This comprehensive genomic characterization provides valuable insights into the local prevalence and evolution of MDR A. baumannii in Bangladesh, reiterating the need for vigilant surveillance to measure the potential spread within the region along with targeted treatment strategies.
ACKNOWLEDGMENTS
This work has been supported by a grant from North South University CTRGC (NSU-CTRG-18-57) to SI.
AUTHOR CONTRIBUTIONS
SF: Investigation; formal analysis; writing – original draft. KMSA: Investigation; formal analysis. AA: Formal analysis; visualization; writing – review & editing. SRS: Formal analysis; writing – review & editing. IJ: Formal analysis; writing – review & editing. KPH: Investigation; formal analysis. AKH: Validation; data curation; formal analysis; visualization. MIK: Data generation. MFAR: Resources; Data generation. MH: writing – review & editing. KNH: writing – review & editing. SI: Conceptualization; methodology; resources; writing – review & editing; supervision; funding acquisition. All authors contributed to the article and approved the submitted version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
SUPPLEMENTARY MATERIALS
Supplementary Table 1. Biofilm formation data, Supplementary Table 2. Pearson correlations between biofilm formation capacity and antibiotic resistance pattern, Supplementary Table 3. The accession number of 50 A. baumannii strains used to construct phylogenomic trees, Supplementary Table 4. The number of accessories, unique, and exclusively absent genes resulted from pan-genome analysis of the twenty different strains of A. baumannii, Supplementary Table 5. Presence of polymorphisms in different antibiotic resistance gene encoding proteins, Supplementary Figure 1. Sequence alignment of five A. baumannii strains, Supplementary Figure 2. Matrix heatmap of the pairwise Average Nucleotide Identity (ANI), and Supplementary Figure 3. Sample figures showing disk diffusion test results for five studied A. baumannii strains against various antibiotics (Supplementary materials).
References
- [1]Murray CJ, Ikuta KS, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet (London, England). 2022;399:629–55.
- [2]Manyi-Loh C, Mamphweli S, et al. Antibiotic Use in Agriculture and Its Consequential Resistance in Environmental Sources: Potential Public Health Implications. Molecules. 2018;23.
- [3]Runa N, Yesmin S, et al. Prevalence of multidrug-resistant ESBL-producing Escherichia coli isolated from beef and sheep meat in Sylhet, Bangladesh. J Adv Biotechnol Exp Ther. 2024;7:520.
- [4]Nusrat T, Akter N, et al. Antibiotic resistance and sensitivity pattern of Metallo-β-Lactamase Producing Gram-Negative Bacilli in ventilator-associated pneumonia in the intensive care unit of a public medical school hospital in Bangladesh. Hosp Pract. 2020:1–9.
- [5]Lee CR, Lee JH, et al. Biology of Acinetobacter baumannii: Pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol. 2017;7.
- [6]Hasan MJ, Rabbani R. The need for adequate research data on carbapenem use and resistance in Bangladesh. Lancet Infect Dis. 2019;19.
- [7]Mia MF, Khan AA, et al. Health Care Associated Multidrug Resistant Acinetobacter Infections in a Tertiary Critical Care Center of Dhaka, Bangladesh. J Bangladesh Coll Physicians Surg. 2020;38:59–63.
- [8]Afroz J, Jubyda FT, et al. Multidrug resistant clinical strains isolated from tracheal aspirates of patients in Dhaka, Bangladesh. Int J Res Med Sci. 2020;8:1094.
- [9]Afroz H, Fakruddin M, et al. Incidence of and risk factors for hospital acquired infection in a tertiary care hospital of Dhaka, Bangladesh. Bangladesh J Med Sci. 2017;16.
- [10]Khan MAS, Chaity SC, et al. Genomic epidemiology of multidrug-resistant clinical Acinetobacter baumannii in Bangladesh. Infect Genet Evol. 2024;123:105656.
- [11]Johnson JS, Spakowicz DJ, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10:1–11.
- [12]CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 31st ed. CLSI supplement M100. Informational Supplement M100-S31. vol. 8. 2021.
- [13]CDC, DGHS. Standard Treatment Guidelines (STG) on Antibiotic Use in Common Infectious Diseases of Bangladesh. Commun Dis Control Dir Gen Heal Serv. 2021;1:1–170.
- [14]Magiorakos AP, Srinivasan A, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81.
- [15]Zhang H, Yu F, et al. Rapid Detection of MCR-Mediated Colistin Resistance in Escherichia coli. Microbiol Spectr. 2022;10.
- [16]Mendoza-Olazarán S, Camacho-Ortiz A, et al. Influence of whole-body washing of critically ill patients with chlorhexidine on Acinetobacter baumannii isolates. Am J Infect Control. 2014;42:874–8.
- [17]Stepanović S, Vuković D, et al. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J Microbiol Methods. 2000;40:175–9.
- [18]Ullah MA, Islam MS, et al. Assessment of prevalence, antibiotic resistance, and virulence profiles of biofilm-forming Enterococcus faecalis isolated from raw seafood in Bangladesh. Heliyon. 2024;10.
- [19]Tabassum J, Anjum A, et al. Draft genome sequencing data of multidrug-resistant Staphylococcus haemolyticus from Bangladeshi hospitals. Data Br. 2024:110918.
- [20]Olson RD, Assaf R, et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023;51:D678–89.
- [21]Wick RR, Judd LM, et al. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595.
- [22]Bankevich A, Nurk S, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19.
- [23]Wick RR, Schultz MB, et al. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31.
- [24]Gurevich A, Saveliev V, et al. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5.
- [25]Li H, Handsaker B, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
- [26]Li W, O’Neill KR, et al. RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021;49:D1020–8.
- [27]Grant JR, Enns E, et al. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023;51:W484–92.
- [28]Alcock BP, Huynh W, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023;51:D690–9.
- [29]Rahman A, Styczynski A, et al. Genomic landscape of prominent XDR Acinetobacter clonal complexes from Dhaka, Bangladesh. BMC Genomics. 2022;23:1–16.
- [30]Jahan MI, Rahaman MM, et al. Draft genome sequence of a carbapenem-resistant clinical Acinetobacter baumannii revealing co-existence of four classes of β-lactamases. J Glob Antimicrob Resist. 2021;27:329–31.
- [31]Diancourt L, Passet V, et al. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One. 2010;5.
- [32]Larsen M V, Cosentino S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–61.
- [33]Siguier P, Perochon J, et al. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34.
- [34]Arndt D, Grant JR, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21.
- [35]Cosentino S, Voldby Larsen M, et al. PathogenFinder - Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS One. 2013;8:e77302.
- [36]Bertelli C, Laird MR, et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45:W30–5.
- [37]Chaudhari NM, Gupta VK, et al. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373.
- [38]Jain C, Rodriguez-R LM, et al. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 2018 91. 2018;9:1–8.
- [39]Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10:2182.
- [40]Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
- [41]Bosi E, Fani R, et al. Defining orthologs and pangenome size metrics. Methods Mol Biol. 2015;1231:191–202.
- [42]Hasan J, Hosen S, et al. The resistance growing trend of common gramnegative bacteria to the potential antibiotics over three consecutive years: a single center experience in Bangladesh. Pharm Pharmacol Int J. 2019;7.
- [43]Liu H, Moran RA, et al. Longitudinal genomics reveals carbapenem-resistant Acinetobacter baumannii population changes with emergence of highly resistant ST164 clone. Nat Commun. 2024;15:9483.
- [44]Sakuma M, Tohya M, et al. Molecular epidemiology of multidrug-resistant Acinetobacter baumannii isolates from a hospital in Nepal. J Glob Antimicrob Resist. 2024;38:363–7.
- [45]Liang J, Zhou K, et al. Identification and Characterization of a Novel Aminoglycoside 3’’-Nucleotidyltransferase, ANT(3’’)-IId, From Acinetobacter lwoffii. Front Microbiol. 2021;12:2487.
- [46]Tahbaz S V., Azimi L, et al. Characterization of aminoglycoside resistance mechanisms in Acinetobacter baumannii isolates from burn wound colonization. Ann Burns Fire Disasters. 2019;32:115.
- [47]Nie L, Lv Y, et al. Genetic basis of high level aminoglycoside resistance in Acinetobacter baumannii from Beijing, China. Acta Pharm Sin B. 2014;4:295–300.
- [48]Biondi S, Long S, et al. Current trends in beta-lactam based beta-lactamases inhibitors. Curr Med Chem. 2011;18.
- [49]Rumbo C, Gato E, et al. Contribution of efflux pumps, porins, and β-lactamases to multidrug resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2013;57:5247–57.
- [50]Lopes BS, Amyes SGB. Insertion sequence disruption of adeR and ciprofloxacin resistance caused by efflux pumps and gyrA and parC mutations in Acinetobacter baumannii. Int J Antimicrob Agents. 2013;41:117–21.
- [51]Park YK, Choi JY, et al. Correlation between overexpression and amino acid substitution of the PmrAB locus and colistin resistance in Acinetobacter baumannii. Int J Antimicrob Agents. 2011;37:525–30.
- [52]Gerson S, Lucaßen K, et al. Diversity of amino acid substitutions in PmrCAB associated with colistin resistance in clinical isolates of Acinetobacter baumannii. Int J Antimicrob Agents. 2020;55.
- [53]Hornsey M, Ellington MJ, et al. AdeABC-mediated efflux and tigecycline MICs for epidemic clones of Acinetobacter baumannii. J Antimicrob Chemother. 2010;65:1589–93.
- [54]Richmond GE, Evans LP, et al. The Acinetobacter baumannii Two-Component System AdeRS Regulates Genes Required for Multidrug Efflux, Biofilm Formation, and Virulence in a Strain-Specific Manner. MBio. 2016;7.
- [55]Atasoy AR, Ciftci IH, et al. Investigation of mutations in adeR and adeS gene regions in gentamicine resistant Acinetobacter baumannii isolates. Biotechnol Biotechnol Equip. 2016;30:360–7.
- [56]Greene C, Wu J, et al. Evaluation of the ability of Acinetobacter baumannii to form biofilms on six different biomedical relevant surfaces. Lett Appl Microbiol. 2016;63:233–9.
- [57]Zeighami H, Valadkhani F, et al. Virulence characteristics of multidrug resistant biofilm forming Acinetobacter baumannii isolated from intensive care unit patients. BMC Infect Dis. 2019;19:1–9.
- [58]Gedefie A, Demsis W, et al. Acinetobacter baumannii Biofilm Formation and Its Role in Disease Pathogenesis: A Review. Infect Drug Resist. 2021;14:3711.
- [59]Karah N, Mateo-Estrada V, et al. The Acinetobacter baumannii website (Ab-web): a multidisciplinary knowledge hub, communication platform, and workspace. FEMS Microbes. 2023;4:1–4.
- [60]Gaillot S, Oueslati S, et al. Genomic characterization of an NDM-9-producing Acinetobacter baumannii clinical isolate and role of Glu152Lys substitution in the enhanced cefiderocol hydrolysis of NDM-9. Front Microbiol. 2023;14:1–11.
- [61]García-González N, Beamud B, et al. Tracking the Emergence and Dissemination of a bla NDM-23 Gene in a Multidrug Resistance Plasmid of Klebsiella pneumoniae . Microbiol Spectr. 2023;11.