Computational analysis of Allium sativum compounds to identify thermolabile hemolysin inhibitors against Vibrio alginolyticus in shrimp
Abstract
Vibrio alginolyticus is one of the major disease-causing bacteria in shrimp aquaculture. The widespread use of antibiotics in shrimp aquaculture to treat bacterial diseases has raised concerns about antibiotic resistance. As a result, alternative treatments, such as plant extract phytochemicals are being explored to mitigate these risks. This study aims to identify promising biologically active compounds from garlic (Allium sativum) that can inhibit the virulent protein thermolabile hemolysin of V. alginolyticus, which causes shrimp vibriosis. Various computational approaches, including molecular docking, pharmacokinetic analysis, and molecular dynamics simulation, were conducted to predict the compounds that can inhibit the phospholipase and hemolysis activities of the thermolabile hemolysin protein. Out of thirty-five compounds from A. sativum, protopine (CID 4970), gibberellin A7 (CID 92782), and gibberellic acid (CID 6466) demonstrated the strongest binding affinities, with scores of -9.4, -8.0, and -7.4 kcal/mol, respectively. Pharmacokinetic and toxicity analyses showed favorable drug-like properties for gibberellin A7 and gibberellic acid with no violations. Molecular dynamics simulations demonstrated that gibberellin A7 and gibberellic acid exhibited the highest stability over 100 nanoseconds. The investigation shows that gibberellin A7 and gibberellic acid from A. sativum have the potential to inhibit the virulent activity of thermolabile hemolysin. However, the study needs further in-vitro and in-vivo analysis to test our predicted results.
INTRODUCTION
Shrimp farming is essential for global food security and has a significant impact on the world economy [1]. It is predicted that by 2050, the global population will exceed nine billion and aquaculture is expected to play a crucial role in meeting the increasing demand for food [2]. In the past decade, global marine shrimp production has expanded and reached over 177,5 million tons in 2019 [3]. Unfortunately, the shrimp farming industry is encountering numerous challenges, such as deteriorating water quality and the infiltration of pathogenic microorganisms [4], resulting in elevated mortality rates and significant economic setbacks that affect global supply and prices [5]. Shrimp vibriosis emerges as a significant bacterial infection triggered by microbes belonging to the Vibrio genus, primarily Vibrio parahaemolyticus, Vibrio alginolyticus and Vibrio vulnificus [6].
V. alginolyticus is a gram-negative, curved rod-shaped bacterium belonging to the family Vibrionaceae [7]. This species is a common inhabitant of marine environments and has been isolated from a variety of aquatic sources, including seawater, sediments, and marine organisms such as fish, shrimp, and shellfish [8]. V. alginolyticus poses a major threat in shrimp aquaculture [9]. Infections of V. alginolyticus is mediated by different virulent factors including, outer membrane protein A (OmpA), outer membrane protein K (OmpK), outer membrane protein U (OmpU), thermostable direct hemolysin (TDH), TDH-related hemolysin (TRH), thermolabile hemolysin (TLH), LuxR, transcriptional regulator (ToxR), and ropS [10].
These virulent proteins of V. alginolyticus cause various types of shrimp diseases, collectively known as shrimp vibriosis, including acute hepatopancreatic necrosis syndrome (AHPNS) [11] and white feces syndrome [12]. The virulent protein TLH known for its hemolytic and phospholipase activities [13]. Thermolabile hemolysin contains a GDSL motif of the esterase-lipase family in its C-terminal domain and exhibits a highly conserved amino acid sequence among Vibrio species [14]. Therefore, TLH is a potential therapeutic target for treating V. alginolyticus in shrimp.
Different groups of antibiotics are largely used in shrimp aquaculture to control Vibrio infection. But the prolonged use of antibiotics reduces their effectiveness and fosters drug resistance in different Vibrio species [10]. Therefore, the need for safer alternatives to antibiotics in aquaculture has become increasingly urgent to ensure its long-term sustainability [15]. The medicinal plant garlic (Allium sativum) contains numerous bioactive compounds [16] with antibacterial properties [17]. It was observed that garlic extracts can prevent shrimp diseases caused by V. parahaemolyticus [18] and V. alginolyticus [19]. Several studies have been conducted to develop potential phytochemical drugs that target the TLH of V. parahaemolyticus [20] and V. harveyi [21], but no studies have been conducted yet for V. alginolyticus. Computer-aided drug design (in silico) is a high-throughput method that accelerates drug discovery by identifying lead compounds faster and at lower costs [22]. Thus, the present study utilized various compounds from A. sativum to identify the TLH activity inhibitors of V. alginolyticus in shrimp using different computational techniques.
MATERIALS AND METHODS
Summary of the methodologies
The whole process is graphically presented in Figure 1.
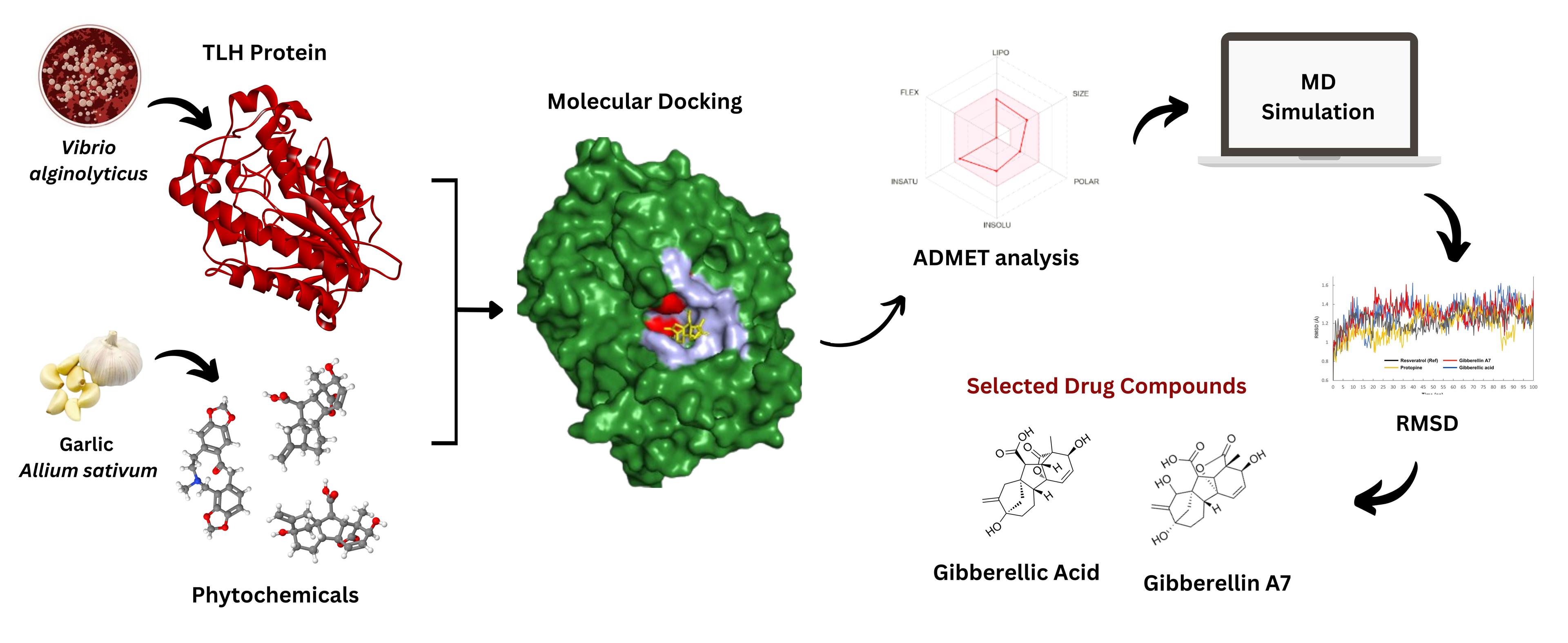
Protein structure retrieval and preparation
The 3D configuration (PDB file) of the virulent TLH protein (PDB ID: 8H09, Resolution: 1.81) [23] was obtained from the RCSB protein data bank [24]. TLH exhibited homodimer chains A and B. Before docking, the protein was prepared and refined using the BIOVIA discovery studio visualizer 4.5 [25]. The protein energy (Hydrogens and heavy atoms) was optimized using Swis-PdbViewer [26]. The ExPASy (Expert Protein Analysis System) [27] database's ProtParam tool was utilized to forecast the physicochemical characteristics of the protein. The Ramachandran plot was checked using PROCHECK to assess the stereochemical quality of the protein structure.
Ligand structure retrieval and preparation
After conducting thorough literature research and utilizing the Indian Medicinal Plant, Phytochemistry, and Therapeutics (IMPPAT) database [28], thirty five compounds of A. sativum bulb were retrieved and saved in a 3D (SDF) file format. Alongside, various medicinal perspectives of these compounds, including anti-fungal, antiviral, antibacterial, and anti-inflammatory properties, were taken into consideration. The compound resveratrol (CID 445254) having antibacterial properties against V. harveyi [21] used as standard control compound.
Molecular docking and interaction study
Before starting molecular docking, the BIOVA discovery studio visualizer 4.5 software was used to predict the active and its associated residues. The virtual screening software PyRx [29] facilitated the molecular docking process of chosen chemical compounds with the receptor protein. The PyRx program's AutoDock Vina wizard [30] was employed to conduct molecular docking, aiming to identify the most favorable binding interactions between our selected target protein and the ligand hit molecules. The target protein's pre-optimized structure was employed as the macromolecule (receptor), while the preprocessed structure of thirty-five phytochemical compounds was utilized as the small molecule (ligand). A grid box was positioned to cover the predicted binding pocket region based on the center and dimension values of the protein. The center of the grid box was X: -24.4630, Y: -35.6537. Z: -24.7282 and the dimensions (Angstrom) were X: 63.4913, Y: 49.7079, Z: 65.9232. The compound with the most negative binding energy (kcal/mol) compared to control resveratrol was chosen for further examination. The most optimal docked outcomes were visualized using the BIOVA Discovery Studio visualizer 4.5 software for subsequent research of protein-ligand interactions.
ADMET analysis
The computational assessment of a molecule's suitability as a drug often involves analyzing its pharmacokinetic (PK) properties, which encompass absorption, distribution, metabolism, and excretion (ADME) [31]. The SwissADME was employed to gather data and evaluate the parameters of the chosen drug candidates [32]. Furthermore, the Lipinski rule of five and bioavailability scores were also considered to assess the drug-likeness of the candidate ligands [33]. Compounds exhibiting undesirable physicochemical properties have been effectively eliminated through the assessment of ADME properties [34].
The toxicity profiling of the selected compounds was performed using the online servers ProTox 3.0 [35], Admet SAR 3.0 [36], and pkCSM [37]. Subsequently, three ligands were selected for further computational studies.
Molecular dynamics simulation study
To assess the stability and behavior of our candidate compounds bound to the target protein, we conducted molecular dynamics (MD) simulations spanning 100 ns. Before simulation, the docked complexes were subjected to energy minimization using the AMBER force field in the YASARA energy minimization module [38]. To simulate the drug-protein complex in a water-based environment, a cubic box was created with periodic boundary conditions, sized at 96.9654 × 96.9654 × 96.9654 Å. The AMBER14 force field was employed for the simulation, and sodium (Na+) and chlorine (Cl-) ions were added using the TIP3P method to maintain system neutrality. Energy minimization of the complex was carried out using the steepest descent method, with van der Waals and short-range Coulomb interactions calculated within an 8 Å radius cut-off. Long-range Coulomb interactions were determined using the PME method. Simulations were conducted under physiological conditions (298 K, pH 7.4, and 0.9% NaCl), running a 100 ns MD simulation with a 2.5 fs time step [39]. Various analyses including root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), and (SASA) were performed.
Binding energy calculation through MM-PBSA
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method is a highly efficient approach for calculating the free energies of various molecular systems [40]. Using the YASARA simulator (YASARA Biosciences, GmbH), the MM-PBSA method was applied to determine the thermodynamic stability of the TLH-resveratrol (control), TLH-protopine, TLH-gibberellin A7, and TLH-gibberellic acid complexes. For these calculations, a 10 ns MD trajectory (from 90 to 100 ns) was derived from the stable range of the TLH-resveratrol (control), TLH-protopine, TLH-gibberellin A7, and TLH-gibberellic acid complexes.
Principal component analysis
To analyze changes in the structural quality of proteins in the presence of ligands during MD simulations, principal component analysis (PCA) was employed to compute various multivariate energy factors [41]. The PCA analysis utilized the final 100 ns of MD trajectory data from four protein-ligand complexes. All computations were performed using Minitab 18 (https://www.minitab.com/en-us/) and custom in-house scripts. For generating plots, the factoextra package was utilized.
RESULTS
Protein structure analysis
The physicochemical properties of the retrieved protein offer valuable insights into its structure, function, and behavior (Table 1). This studied protein chain is composed of 418 amino acids, with a molecular weight determined to be 47318.89 Daltons. These amino acids are estimated to have a half-life of approximately 10 hours. The isoelectric point (pI) at pH 5.14 indicates their charge-neutral condition. An aliphatic index (AI) of 68.73 reflects the thermostability of the protein. Moreover, the instability index (II) of 29.92 indicates significant stability. The grand average of hydropathicity (GRAVY) is -0.382, suggesting an average hydrophilic character. The extracellular GDSL lipases family proteins demonstrate hydrolase activity, specifically targeting ester bonds. In addition, the Ramachandran plot shows 90.2% of residues in the most favored regions, indicating that the structure is sufficiently accurate for further docking analysis.
Table 1. Properties of thermolabile hemolysin protein predicted by ExPASy server.
Molecular docking and interaction analysis
The three-dimensional structure of TLH (PDB ID: 8H09; R-Value Free: 0.204) and thirty-five compounds from A. sativum were utilized for docking. The 35 compounds were ranked according to their docking scores (Supplementary Table 1). Compounds with docking scores lower than the control drug Resveratrol (-7.4 Kcal/mol) were selected for further pharmacodynamic and simulation analysis. Among the 35 compounds, three compounds, Protopine (-9.4 Kcal/mol), Gibberellin A7 (-8.0 Kcal/mol), and Gibberellic acid (-7.4 Kcal/mol) exhibited the highest binding affinities, compared to the control one (Table 2).
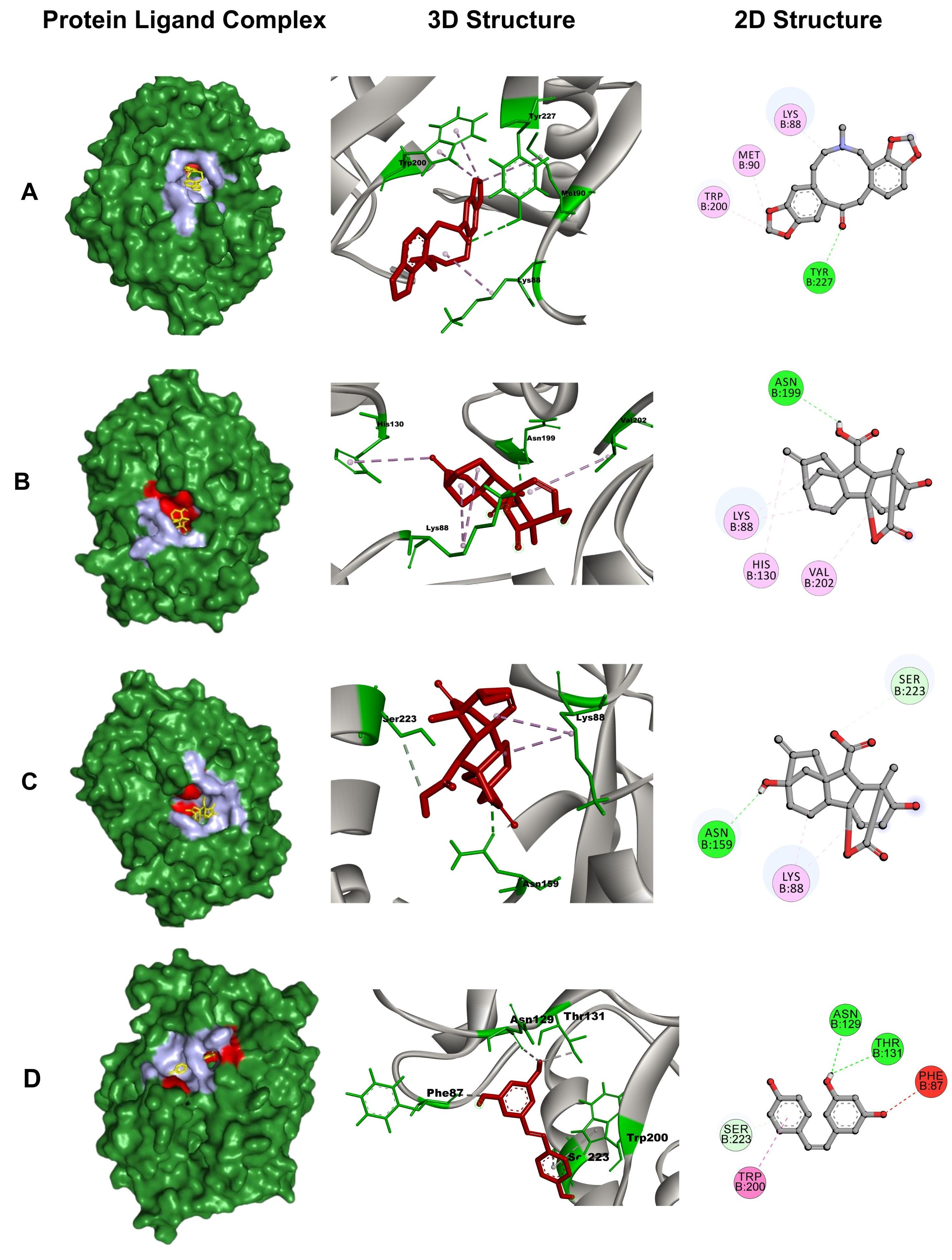
Protein-ligand bonds are crucial for drug binding and stabilizing drug-protein complexes. Hydrophobic interactions, hydrogen bonds, and electrostatic interactions at varying distances play crucial roles in determining ligand binding affinities. Table 3 provides detail and Figure 2 visually represents the binding conformations of these compounds within the TLH common binding site. Protopine (CID 4970) displayed one conventional hydrogen bond interaction with the residue Tyr227 (2.85 Å) and three hydrophobic interactions (p-alkyl and alkyl) with Met90 (4.79 Å), Lys88 (4.23 Å), and Trp200 (4.69 Å, 4.48 Å). Gibberellin A7 (CID 92782) exhibited one conventional hydrogen bond with Asn199 (2.80 Å) and three hydrophobic interactions (p-alkyl and alkyl) with Lys88 (4.93 Å, 5.22 Å), Val202 (5.21 Å), and His130 (5.05 Å). Gibberellic acid (CID 6466) demonstrated one conventional hydrogen bond with Asn159 (2.64 Å), one carbon-hydrogen bond with Ser223 (3.79 Å), and one hydrophobic (alkyl) interaction with Lys88 (3.68 Å, 4.44 Å).
Table 2. Identity and binding affinity of the top three compounds from A. sativum with control resveratrol.
Table 3. Bonding interaction of Resveratrol, Protopine, Gibberellic acid, and Gibberellin A7 with TLH.
ADMET analysis
The ADME analysis assesses both the physicochemical properties and biological functions of the compound, as well as its drug-likeness. The physicochemical properties of the three highest docked compounds extracted from A. sativum bulb were evaluated using Lipinski’s rule. This rule sets criteria including a molecular weight (MW) below 500 Da, lipophilicity (log P) under 5, and a maximum of 10 hydrogen bond acceptors (HBA) and 5 hydrogen bond donors (HBD) to determine drug-likeness. It is apparent that protopine, gibberellin A7, and gibberellic acid all meet Lipinski’s rule criteria (drug likeliness: 0 violation) (Table 4).
The toxicological properties of the three molecules were predicted using the AdmetSAR 3.0 and ProTox-III servers (Table 5). The results showed that none of the compounds were carcinogenic, hepatotoxic, or cytotoxic, but protopine exhibited active immunogenicity. Additionally, all substances had higher LD50 values, except protopine (950 mg/kg) indicating their relative safety for pharmaceutical use as they are less likely to cause adverse effects.
Table 4. Physicochemical properties and pharmacokinetic predictions of the top 3 selected compounds of the study by SwissADME.
Table 5. Toxicity properties analysis of selected top three ligands’ through Admet SAR and Protox-III online server.
Molecular dynamics simulation analysis
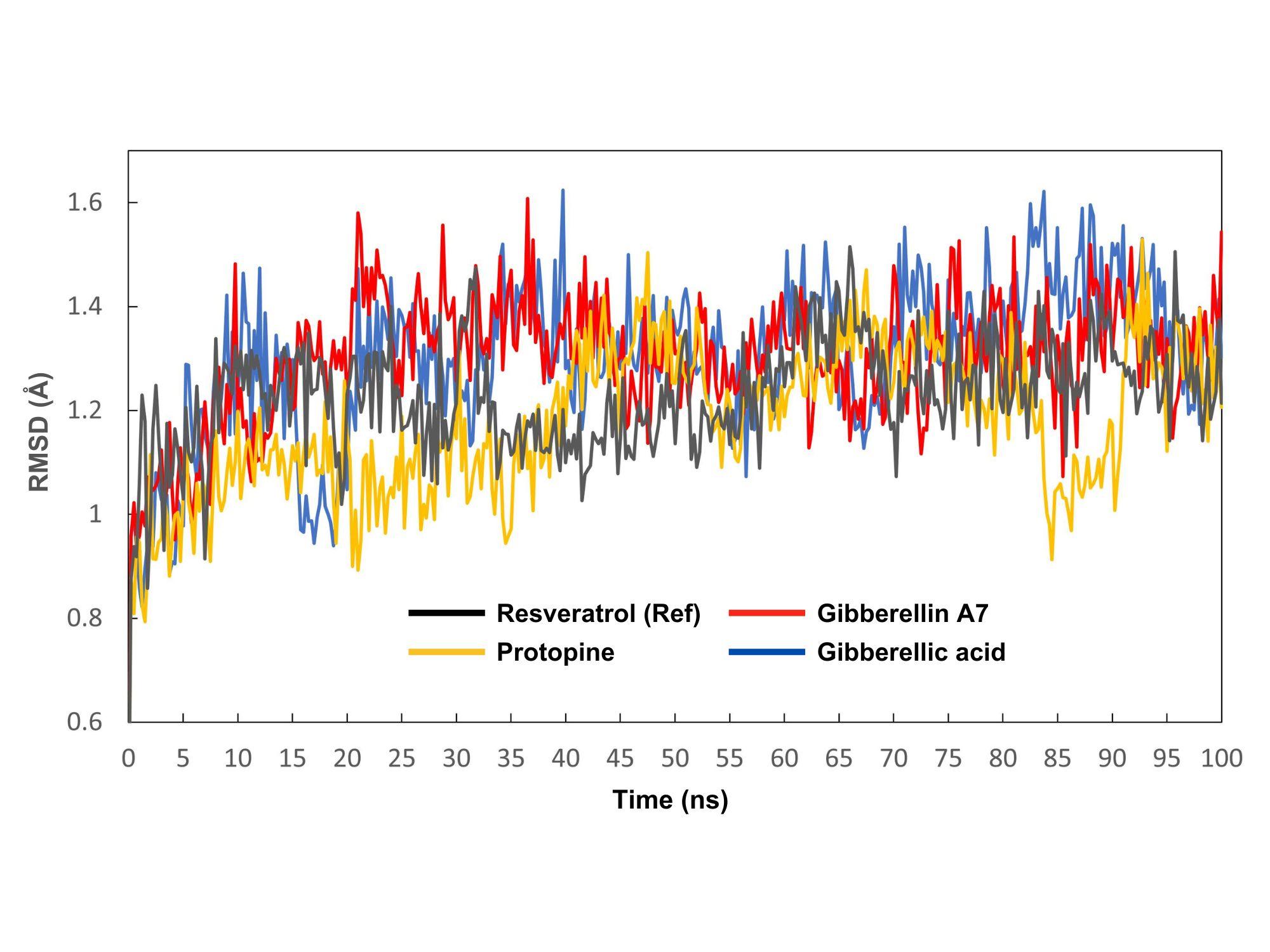
The MD simulation trajectories generated by the Yasara software were used to analyze RMSD, RMSF, Rg, and SASA values. The control compound resveratrol was utilized to observe the fluctuations between complexes. RMSD was employed to assess structural reliability and identify conformational changes. In Figure 3, the protein-ligand complexes of resveratrol (control), protopine, gibberellin A7 and gibberellic acid were visualized in gray, yellow, red, and blue colors, respectively. The average RMSD values of resveratrol (control), protopine, gibberellin A7 and gibberellic acid are 1.22 Å, 1.18 Å, 1.29 Å, and 1.30 Å, respectively. Tested compounds protopine, gibberellin A7, and gibberellic acid consistently maintained stability alongside the standard compound resveratrol throughout the 0-100 ns timeframe.
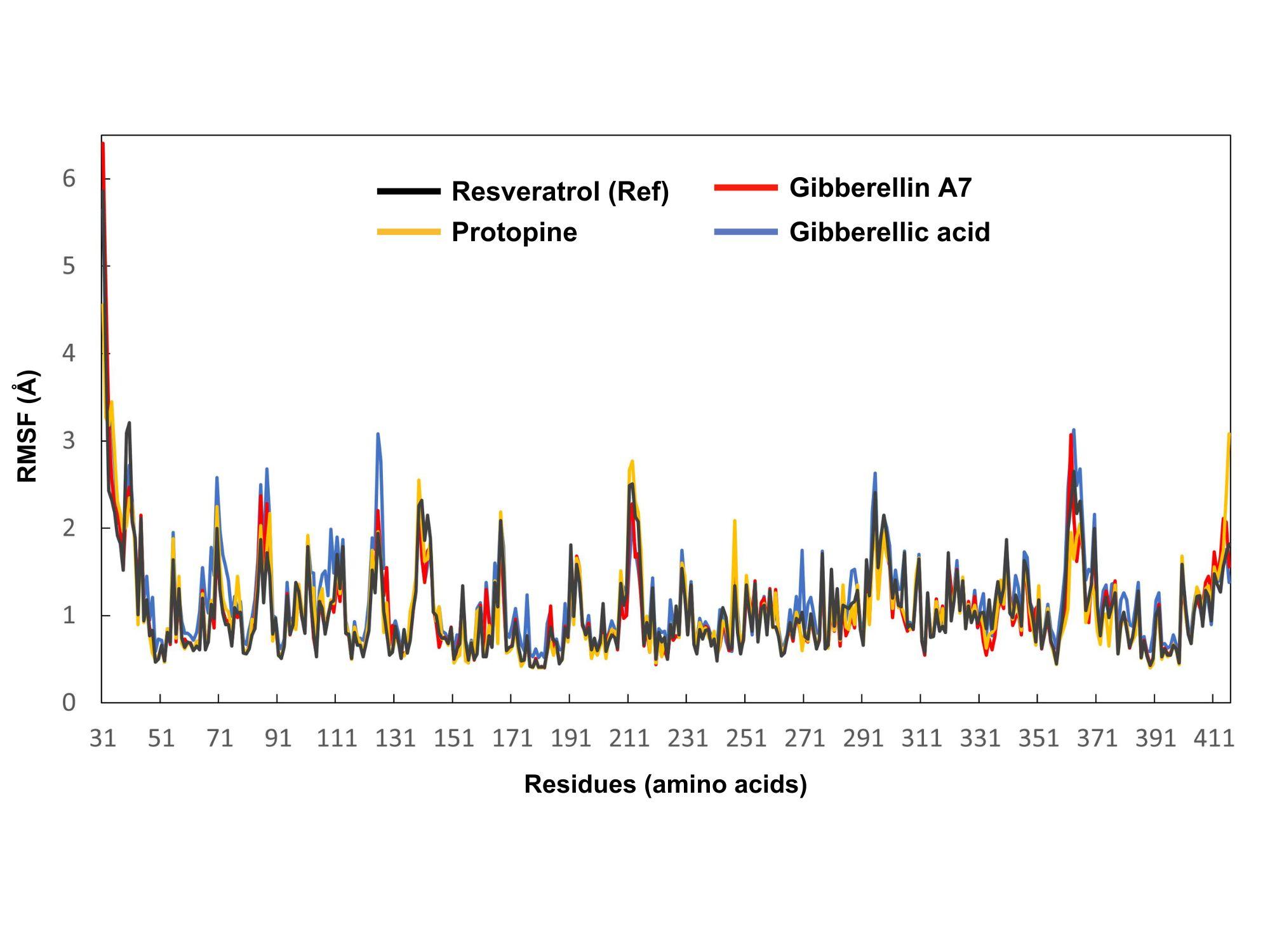
The stability of each amino acid residue in the thermolabile hemolysin backbone was analyzed through RMSF by measuring its fluctuations when bound to resveratrol (control), protopine, gibberellin A7, and gibberellic acid. Residues of the protein that interact with the ligands are shown in Figure 4. The RMSF values of resveratrol (control), protopine, gibberellin A7, and gibberellic acid protein-ligand complexes were visualized in gray, yellow, red, and blue colors respectively. The average residual fluctuations for the resveratrol, protopine, gibberellin A7, and gibberellic acid complexes were found 1.07, 1.06, 1.05, and 1.18, respectively. The peaks on the graph represent regions of the protein that exhibit the most significant fluctuations throughout the simulation period. Protopine had the highest fluctuation at residue ILE 212 (2.77 Å) and the lowest at HIS 178, SER 180, and ASN 181 (0.4 Å each). Gibberellin A7 exhibited its highest fluctuation at residue ARG 362 (3.07 Å) and the lowest at SER 180 and GLY 182 (0.4 Å each). Gibberellic Acid showed the highest fluctuation at residue SER 363 (3.13 Å) and the lowest at HIS 399 (0.51 Å).
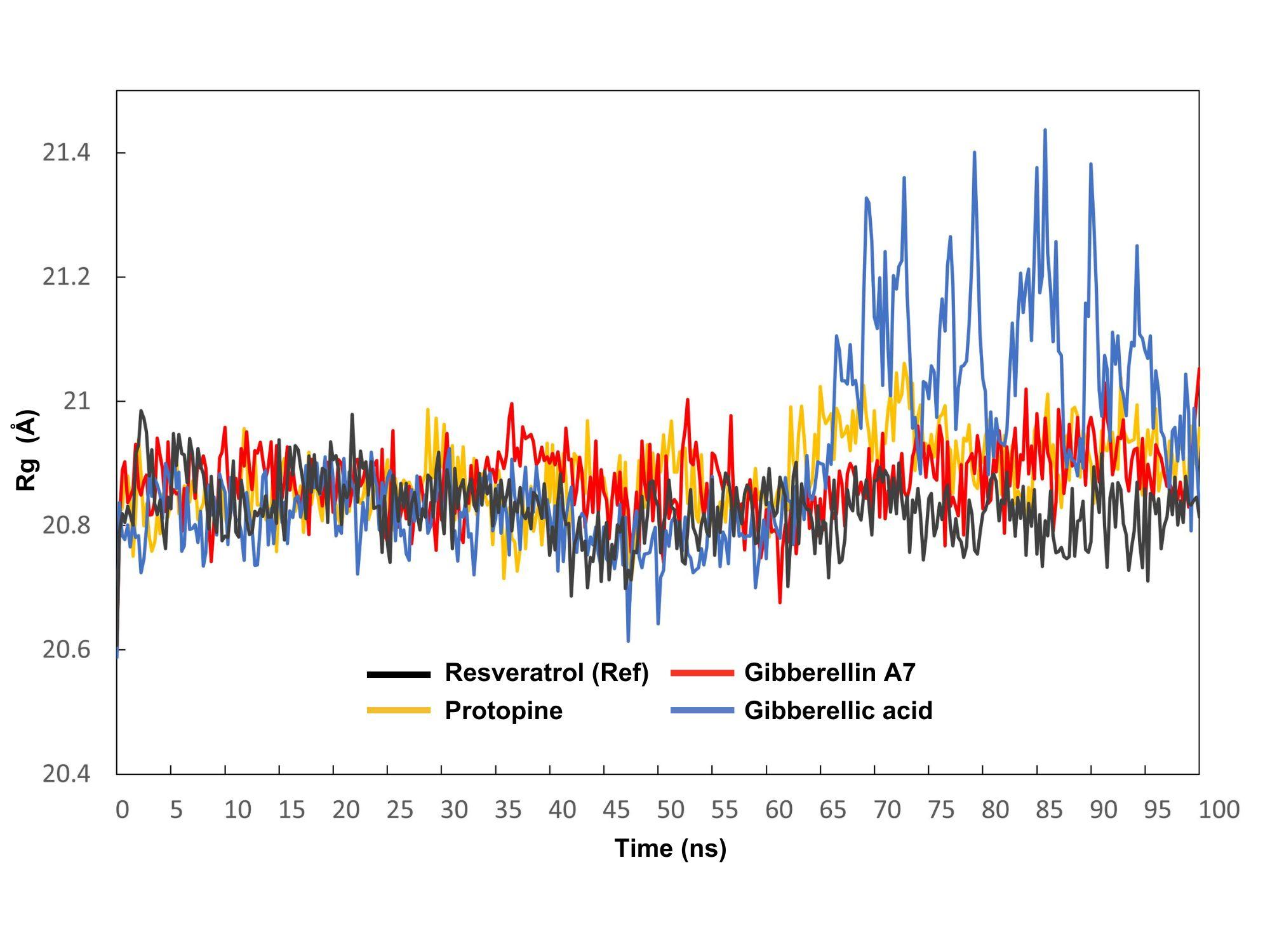
The radius of gyration (Rg) represents the distance between the center of mass of the atoms in a protein and its overall structure over time, providing insight into its compactness and stability. The average Rg values for the resveratrol (control), protopine, gibberellin A7, and gibberellic acid complexes were observed 0.39 Å, 0.35 Å, 0.37 Å, and 0.83 Å, respectively (Figure 5). For protopine, the Rg fluctuated between a maximum of 21.061 Å at 72.75 ns and a minimum of 20.711 Å at 47.5 ns. Gibberellin A7 had a maximum Rg of 21.053 Å at 100 ns and a minimum of 20.676 Å at 61.25 ns, while gibberellic acid exhibited a maximum Rg of 21.437 Å at 85.75 ns and a minimum of 20.614 Å at 47.25 ns.
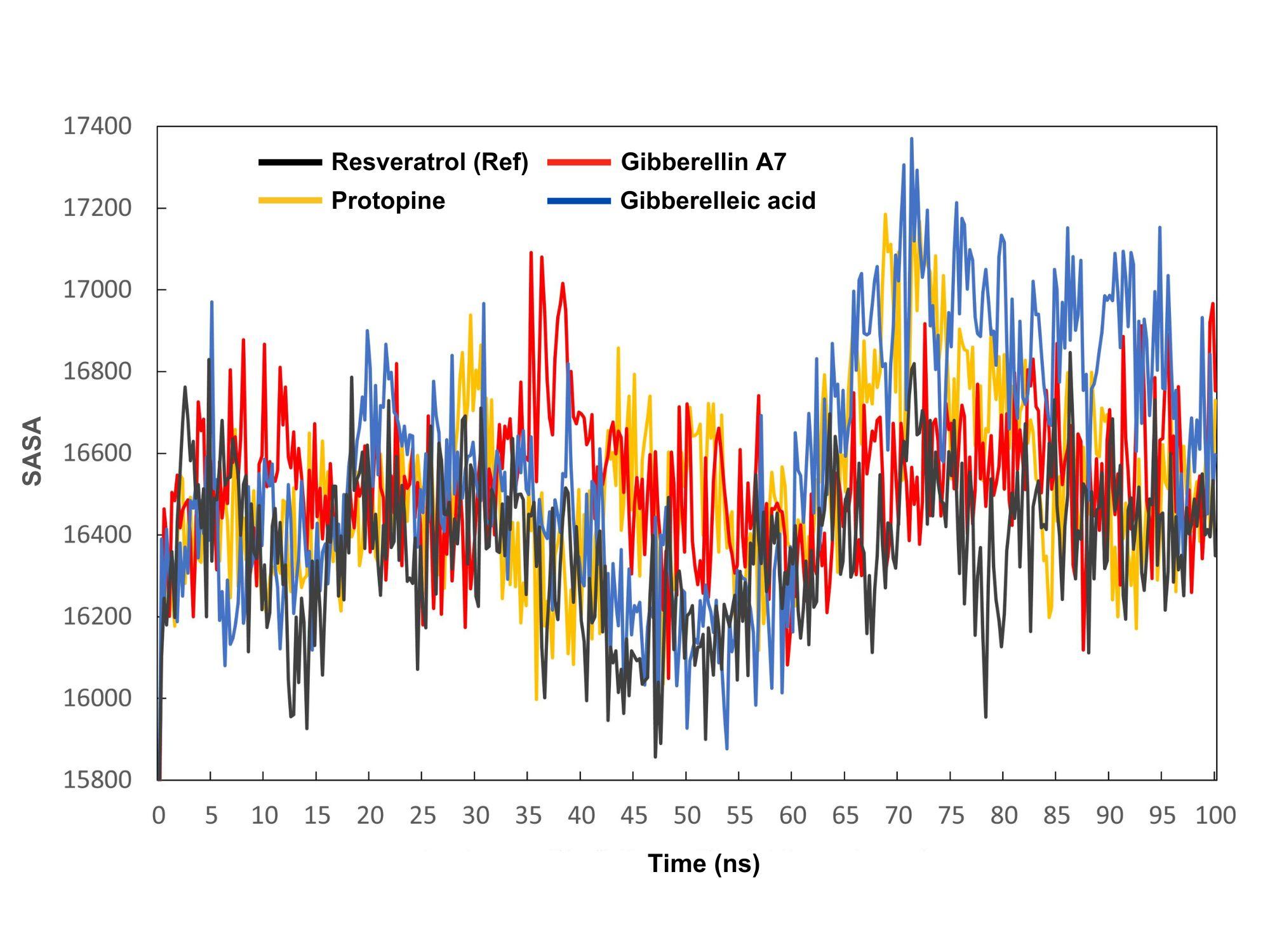
The solvent-accessible surface area (SASA) value shows how accessible a molecule's surface is to solvent molecules. SASA measures molecular hydrophilicity or hydrophobicity, which affects solubility and permeability. Figure 6 shows all complexes' SASA values from 0 to 100 ns. The average SASA values varied among substances, with resveratrol (control), protopine, gibberellin A7, and gibberellic acid averaging 16367.82, 16513.48, 16525.87, and 16573.17 Ų, respectively. High SASA values indicate protein expansion and low levels signify truncation.
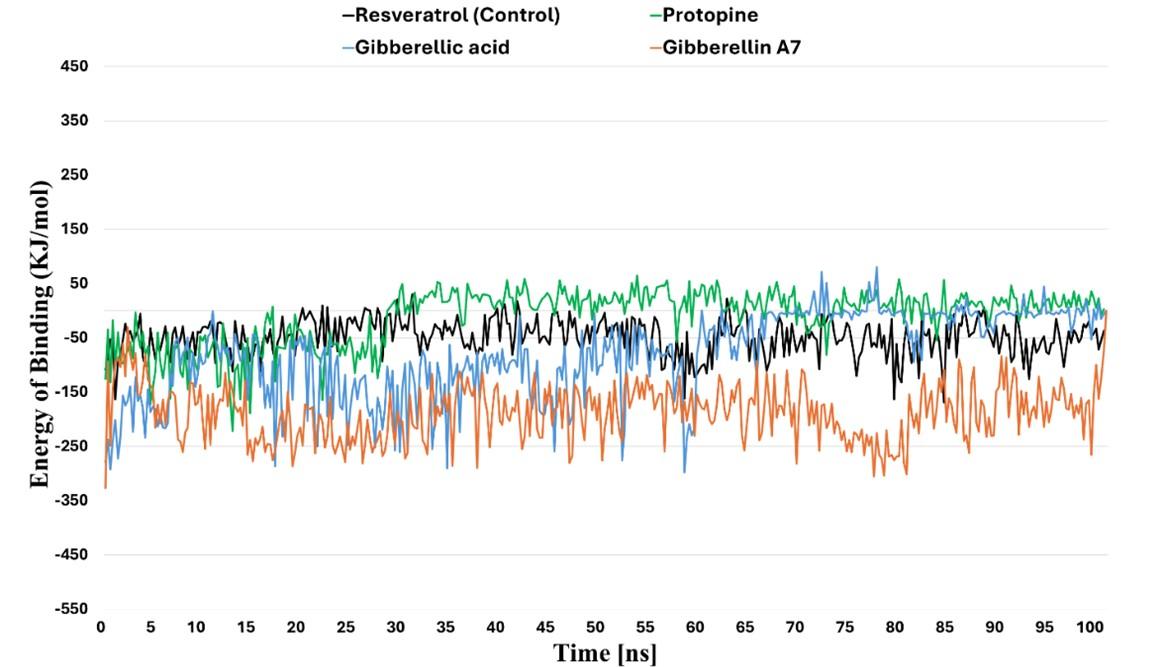
MM/PBSA analysis
Using the MM/PBSA method in the YASARA stimulator, the binding free energy of the three compounds to TLH was calculated quantitatively. This analysis aims to estimate the average binding energy per 0.25 ns interval of TLH-Resveratrol (control), TLH-protopine, TLH-gibberelin A7, TLH-gibberelic acid: -48.647 KJ/mol, -10.707 KJ/mol, -188.18 KJ/mol, -81.585 KJ/mol, respectively (Figure 7). According to MM/PBSA analysis, the three above compounds bind to TLH with a significant binding affinity and form a stable complex compared to the control, since the control showed less negative binding energy.
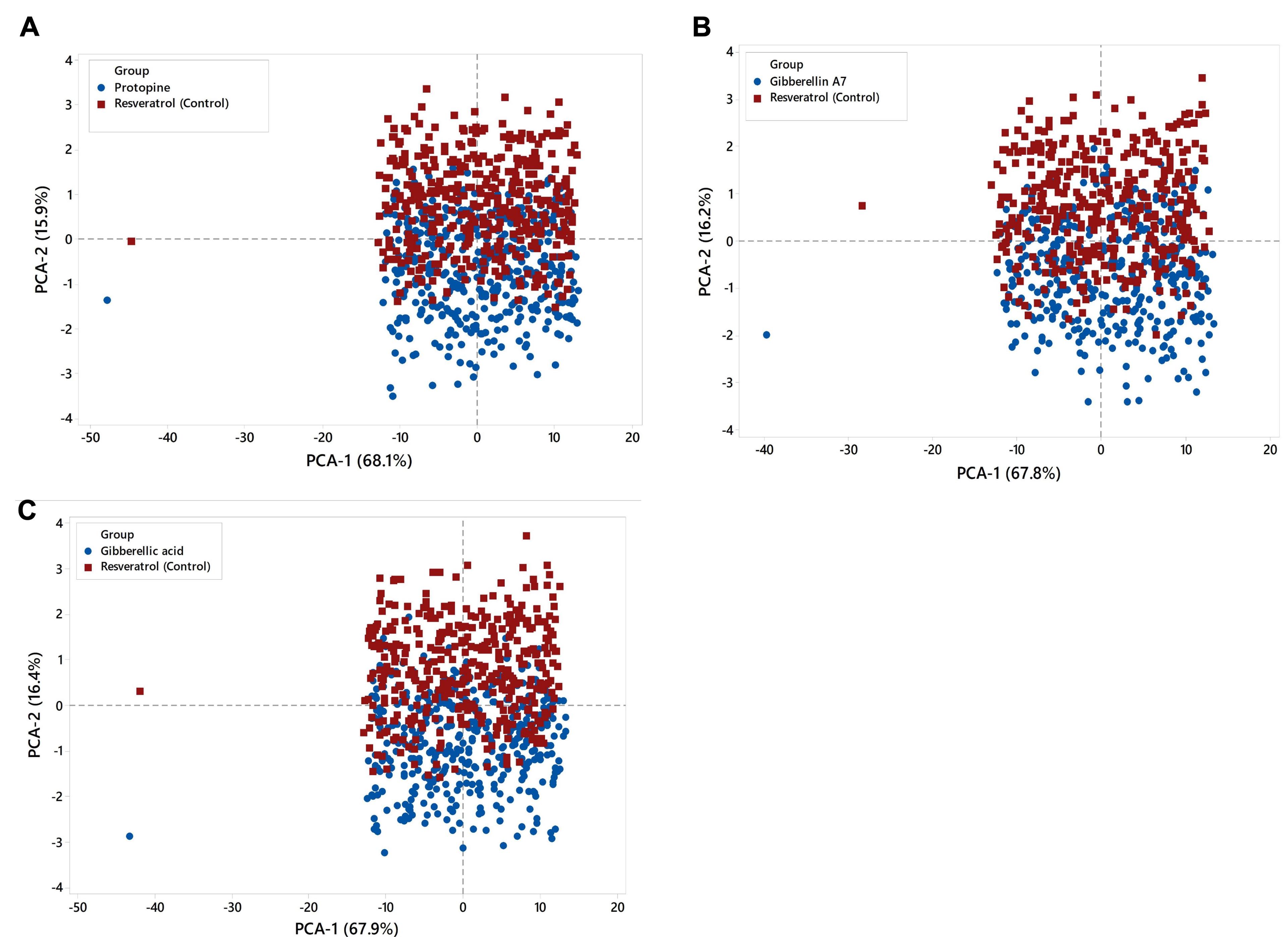
Principal component analysis
Principal component analysis (PCA) was used to predict significant coordinated motions during ligand binding. Figure 8 shows PCA cluster distributions on structural and energy factors. Each dot represents a conformer, representing MD simulation's structural and energetic changes. In the PCA model Figure 8A, PC1 and PC2 account for 84%, with PC1 contributing 68.1% and PC2 15.9%. PC1 and PC2 account for 84% of PCA model Figure 8B, with PC1 contributing 67.8% and PC2 16.2%. PCA model Figure 8C shows PC1 and PC2 explain 84.3%, with PC1 contributing 67.9% and PC2 16.4%. The score plot reveals control chemical complexes overlap the protein. The PCA loading plot shows that the complex is positively correlated with bond, angle, and Van der Waals (VdW) variables.
DISCUSSION
The physicochemical properties of a protein are crucial factors in drug design and drug target prediction. TLH (PDB ID: 8H09) from V. alginolyticus shows the highest resolution (1.81 Å) and highest reliability [42]. Thus, the aliphatic index (AI) of 68.73 for the studied protein suggests thermal stability and a high content of hydrophobic amino acids, making it a potentially attractive drug target due to its ability to maintain structural integrity in diverse physiological conditions [43]. Proteins with higher stability may be more attractive drug targets as they are less likely to undergo conformational changes or denaturation during binding with drugs [44]. Moreover, the instability index (II) (29.92) of the studied protein suggests its stability, as proteins with instability index values below forty are considered stable [45]. The negative GRAVY value (-0.382) indicates the protein's hydrophilic nature, which is vital for facilitating its interaction with water molecules and ensuring solubility [46]. In addition, the extracellular presence of the TLH protein enhances its drug ability and makes it relevant to disease and drug discovery. Additionally, the GDSL lipase domain, coupled with its phospholipase and hemolytic activity, contributes to disease in the host body [47]. Nonetheless, former studies found V. alginolyticus TLH induced apoptosis and necrosis in silver sea bream (Rhabdosargus sarba) [48]. Moreover, their enzymes hemolytic and phospholipase activity were observed in V. parahaemolyticus [49] and V. harveyi infection [50]. Therefore, targeting this domain and inhibiting its function may reduce V. alginolyticus infection in shrimp.
The molecular docking results filter the top three compounds with the highest docking scores (negative values): protopine (-9.4), gibberellin A7 (-8.0), and gibberellic acid (-7.4). The lowest docking score reflects the highest binding affinity, implying that the compound with the lowest score forms a more stable complex and maintains longer contact [51]. In this investigation, the molecular docking results indicate that these three compounds interact at the TLH active binding site. Hydrogen bond analysis revealed that both gibberellin A7 and gibberellic acid form hydrogen and hydrophobic bonds with Asn 159 and Lys88 amino acid residues.
The three compounds with the highest docking scores and stable in MD simulation were analyzed for ADMET parameters using the online tool SwissADME and all of them exhibited favorable ADMET properties. Mostly, ADMET evaluates drug pharmacokinetics (PK), and optimizing PK parameters is crucial before progressing to a potential drug candidate to meet standard clinical trial requirements [52]. The selected compounds exhibit drug-like characteristics as they meet Lipinski’s Rule of Five (RO5), which considers factors like molecular weight (<500 Daltons), lipophilicity (LogP < 5), and hydrogen bonding properties [33]. However, adherence to Lipinski's rule doesn't ensure drug potency. Additionally, according to Veber's rule [53], orally bioavailable drugs usually have fewer than 10 rotatable bonds and a topological polar surface area (TPSA) value of less than 140. Analysis of this study revealed that all three compounds, protopine, gibberellin A7, and gibberellic acid have fewer than 10 rotatable bonds and TPSA values of 57.23, 104.06, and 83.83, respectively, all below 140. Examining the toxicology profiles of drug candidates offers insights into potential risks to shrimp and the environment. The LD50 (median lethal dose) assesses a substance's lethal toxicity, with a higher value (>5000 mg/kg) indicating lower toxicity and a lower value (<5 mg/kg) implying higher toxicity [54]. The compound protopine at 940 mg/kg is classified as slightly toxic and is a concern compound as a therapeutic agent, while gibberellin A7 and gibberellic acid at 6300 mg/kg are considered partially non-toxic. Thus, predictions from the ProTox and admetSAR web server ensure a safe selection of stable compounds as potential drugs [35, 36].
Molecular dynamics simulation revealed that the RMSD and RMSF values of the complexes ranged between 1.18 to 1.32 Å and 1.05 to 1.18 Å, respectively. These results indicate the stability of these interactions within the acceptable RMSD and RMSF range of 0.01 to 3.5 Å. RMSD and RMSF values ideally fall within the range of 1-3 Å, indicating stability. The tested compounds protopine, gibberellin A7, and gibberellic acid showed stability similar to the control resveratrol (RMSD 1.22 Å) over the 0-100 ns simulation. However, RMSF analysis revealed gibberellic acid had the highest residue fluctuation (3.13 Å at SER 363), while resveratrol displayed more stability with an average RMSF of 1.07 Å, indicating resveratrol binding resulted in less flexibility and greater stability in the thermolabile hemolysin backbone. The Radius of gyration (Rg) showed a narrow range of 0.35 to 0.83 Å, significantly below the maximum of 1.50, indicating excellent complex stability. The maximum range of Rg can be 1.50 to ensure complex stability. None of the compounds, including the control, demonstrated structural switching during the simulation, indicating a stable protein-ligand complex throughout. The SASA values >15000 also show the stability of these compounds. The SASA analysis indicated greater surface exposure and potential for protein expansion compared to standard resveratrol.
Finally, gibberellin A7 and gibberellic acid were identified as potential compounds, while protopine was excluded due to its toxic properties. These two compounds of garlic have antibacterial properties. Garlic (A. sativum) contains various bioactive compounds known for their beneficial properties for aquatic animal health [17]. Gibberellin A7 and gibberellic acid are plant-derived phytohormones that have antimicrobial activity and regulate plant development and growth. A previous study revealed that in modern aquaculture, natural plant products such as flavonoids, alkaloids, terpenoids, and saponins replace the chemical compounds and antibiotics [55]. Specifically, phenolic compounds have been identified as inhibitors of the TLH produced by V. parahaemolyticus, demonstrating their potential for combating bacterial virulence [20]. Hannan et al. 2009 demonstrated that A. sativum inhibited the growth of V. alginolyticus, the bacteria responsible for shrimp vibriosis, in in vitro conditions at a dose of 10 mg/kg, consistent with our findings [19]. In addition, the used control drug resveratrol is a polyphenol with antioxidant properties that negatively regulates the transcription level and inhibits the hemolysin activity of V. harveyi [21]. Overall, the results of the present study indicate that the two chosen compounds exhibit favorable binding and stability during their interaction. These phytochemicals have the potential to be developed as antibacterial drugs against V. alginolyticus in shrimp, but further in vitro and in vivo studies are needed for drug development.
CONCLUSIONS
In aquaculture, shrimp infections caused by V. alginolyticus present a significant challenge. Traditional treatments such as antibiotics and chemicals have drawbacks, including the promotion of antibiotic resistance and high costs. Garlic (A. sativum) extracts, known for their antimicrobial properties, are particularly effective in mitigating shrimp Vibrio infections. In this study, a diverse bioinformatics approach, including virtual screening, pharmacological analysis, and simulation studies was employed to identify potential inhibitors against bacterial (V. alginolyticus) infection in shrimp. These computational approaches exhibited the possibility of a strong inhibitory potential of two promising compounds such as gibberellin A7 and gibberellic acid, in disrupting the TLH activity of V. alginolyticus in shrimp. Further studies are needed to validate the result of this study.
ACKNOWLEDGEMENTS
None.
AUTHOR CONTRIBUTIONS
SMB: Conceptualization, Methodology, Software, Formal Analysis, Visualization, Writing - Original Draft; NBR: Software, Formal Analysis, Writing - Original Draft. MM: Data Curation, Software; SFA: Software, Formal Analysis; MAH: Writing – Interpretation, Review & Editing; MNJ: Resources, Review & Editing; MAH: Resources, Review & Editing; MM: Writing - Review & Editing; MSA: Writing - Review & Editing. All authors have read and agreed to the published version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
SUPPLEMENTARY MATERIALS
Supplementary Table 1. Docking scores of 35 selected compounds. This table illustrates the 35 compounds of Allium sativum with their identity, chemical name, molecular weight, chemical formula, two-dimensional structure, and molecular docking scores (Supplementary materials).
References
- [1]Azra MN, Okomoda VT, et al. The contributions of shellfish aquaculture to global food security: Assessing its characteristics from a future food perspective. Front Mar Sci. 2021; 8:654897.
- [2]Godfray HCJ, Beddington JR, et al. Food security: The challenge of feeding 9 billion people. Science. 2010; 327:812-818.
- [3]Lichna AI, Bezyk КІ, et al. Analysis of FAO data on the global fisheries and aquaculture production volume. Водні біоресурси. 2023; 1(13):188-97.
- [4]Gunalan B, Soundarapandian P, Anand T, Kotiya AS, Simon NT. Disease Occurrence in Litopenaeus vannamei shrimp culture systems in different geographical regions of India. Int J Aquaculture. 2014; 4(4):24-27.
- [5]Asche F, Anderson JL, et al. The economics of shrimp disease. J Invertebr Pathol. 2021; 186:107397.
- [6]Abdel-Latif HMR, Yilmaz E, et al. Shrimp vibriosis and possible control measures using probiotics, postbiotics, prebiotics, and synbiotics: A review. Aquaculture. 2022; 551:737951.
- [7]Sampaio A, Silva V, et al. Vibrio spp.: Life strategies, ecology, and risks in a changing environment. Diversity. 2022; 14:97.
- [8]Triga A, Smyrli M, et al. Pathogenic and opportunistic Vibrio spp. associated with vibriosis incidences in the Greek aquaculture: The role of Vibrio harveyi as the principal cause of vibriosis. Microorganisms. 2023; 11:1197.
- [9]de Souza Valente C, Wan AHL. Vibrio and major commercially important vibriosis diseases in decapod crustaceans. J Invertebr Pathol. 2021; 181:107527.
- [10]Yu Y, Li H, et al. Antibiotic resistance, virulence and genetic characteristics of Vibrio alginolyticus isolates from aquatic environment in costal mariculture areas in China. Mar Pollut Bull. 2022; 185:114219.
- [11]Tran L, Nunan L, et al. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis Aquat Organ. 2013; 105:45-55.
- [12]Kumara KRPS. White faeces syndrome caused by Vibrio alginolyticus and Vibrio fluvialis in shrimp, Penaeus monodon (Fabricius 1798) - multimodal strategy to control the syndrome in Sri Lankan grow-out ponds. Asian Fish Sci. 2017; 30(4):246-261
- [13]Wan Y, Liu C, et al. Structural analysis of a Vibrio phospholipase reveals an unusual Ser-His-chloride catalytic triad. J Biol Chem. 2019; 294(30):11391-11401.
- [14]Akoh CC, Lee GC, et al. GDSL family of serine esterases/lipases. Prog Lipid Res. 2004; 43:534-552.
- [15]Bondad-Reantaso MG, MacKinnon B, et al. Review of alternatives to antibiotic use in aquaculture. Rev Aquac. 2023; 15:1421-1451.
- [16]Nakamoto M, Kunimura K, et al. Antimicrobial properties of hydrophobic compounds in garlic: allicin, vinyldithiin, ajoene and diallyl polysulfides. Exp Ther Med. 2019; 19:1550-1553.
- [17]Valenzuela-Gutiérrez R, Lago-Lestón A, et al. Exploring the garlic (Allium sativum) properties for fish aquaculture. Fish Physiol Biochem. 2021; 47:1179-1198.
- [18]Ramadhaniah V, Indrawati A, et al. Activity of garlic (Allium Sativum L.) extract against Vibrio parahaemolyticus bacteria. IOP Conf Ser Earth Environ Sci. 2023; 1174:012004.
- [19]Hannan MDA, Rahman M, et al. Molecular identification of Vibrio alginolyticus causing vibriosis in shrimp and its herbal remedy. Pol J Microbiol. 2019; 68:429-438.
- [20]Vazquez-Morado LE, Robles-Zepeda RE, et al. Biochemical characterization and inhibition of thermolabile hemolysin from Vibrio parahaemolyticus by phenolic compounds. PeerJ. 2021; 9:e10506.
- [21]Zhao X, Guo Y, et al. Resveratrol inhibits the virulence of Vibrio harveyi by reducing the activity of Vibrio harveyi hemolysin. Aquaculture. 2020; 522:735086.
- [22]Jabalia N, Kumar A, et al. In silico approach in drug design and drug discovery: an update. Innovations and Implementations of Computer Aided Drug Discovery Strategies in Rational Drug Design. Springer: New York, NY, 2021. pp 245-271.
- [23]Wang C, Liu C, et al. Catalytic site flexibility facilitates the substrate and catalytic promiscuity of Vibrio dual lipase/transferase. Nat Commun. 2023; 14:1-11.
- [24]Rose PW, Bi C, et al. The RCSB protein data bank: new resources for research and education. Nucleic Acids Res. 2013; 41:D475-D482.
- [25]Design LI G A N D. Pharmacophore and ligand-based design with Biovia Discovery Studio®. BIOVIA: California, 2014.
- [26]Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View). Brief Bioinform. 2001; 2:195-197.
- [27]Gasteiger E, Hoogland C, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005: 571-607.
- [28]Mohanraj K, Karthikeyan BS, et al. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci Rep. 2018; 8:1-17.
- [29]Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015; 1263:243-250.
- [30]Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31:455-461.
- [31]Guan L, Yang H, et al. ADMET - score - a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2018; 10:148-157.
- [32]Daina A, Michielin O, et al. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017; 7:1-13.
- [33]Lipinski CA, Lombardo F, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001; 46:3-26.
- [34]Martínez-Archundia M, Bello M, et al. Design of drugs by filtering through ADMET, physicochemical and ligand-target flexibility properties. Rational Drug Design: Methods and Protocols. Springer: New York, NY, 2018, pp 403-416.
- [35]Banerjee P, Eckert AO, et al. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018; 46:W257-W263.
- [36]Yang H, Lou C, et al. AdmetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019; 35:1067–1069.
- [37]Pires DE, Blundell TL, et al. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of medicinal chemistry. 2015; 58(9):4066-4072.
- [38]Michaud-Agrawal N, Denning EJ, et al. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011; 32:2319-2327.
- [39]Kumar SP, Patel CN, et al. Molecular dynamics-assisted pharmacophore modeling of caspase-3-isatin sulfonamide complex: Recognizing essential intermolecular contacts and features of sulfonamide inhibitor class for caspase-3 binding. Comput Biol Chem. 2017; 71:117-128.
- [40]Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol Informatics. 2012; 31:114-122.
- [41]Giuliani A. The application of principal component analysis to drug discovery and biomedical data. Drug Discov Today. 2017; 22:1069-1076.
- [42]Thomson RES, Carrera-Pacheco SE, et al. Engineering functional thermostable proteins using ancestral sequence reconstruction. J Biol Chem. 2022; 298:102435.
- [43]Akbarian M, Chen SH. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics. 2022; 14:2533.
- [44]Gamage DG, Gunaratne A, et al. Applicability of Instability Index for in vitro protein stability prediction. Protein Pept Lett. 2019; 26:339–347.
- [45]Efremov RG, Chugunov AO, et al. Molecular lipophilicity in protein modeling and drug design. Curr Med Chem. 2007; 14:393–415.
- [46]Huxley-Jones J, Foord SM, et al. Drug discovery in the extracellular matrix. Drug Discov Today. 2008; 13:685–694.
- [47]Jia A, Woo NYS, et al. Expression, purification, and characterization of thermolabile hemolysin (TLH) from Vibrio alginolyticus. Dis Aquat Org. 2010; 90:121-127.
- [48]Wong SK, Zhang XH, et al. Vibrio alginolyticus thermolabile hemolysin (TLH) induces apoptosis, membrane vesiculation and necrosis in sea bream erythrocytes. Aquaculture. 2012; 330–333:29-36.
- [49]Shinoda S, Matsuoka H, et al. Purification and characterization of a lecithin-dependent haemolysin from Escherichia coli transformed by a Vibrio parahaemolyticus gene. Microbiology. 1991; 137:2705-2711.
- [50]Zhong Y, Zhang XH, et al. Overexpression, purification, characterization, and pathogenicity of Vibrio harveyi hemolysin VHH. Infect Immun. 2006; 74:6001–6005.
- [51]Torres PHM, Sodero ACR, et al. Key topics in molecular docking for drug design. Int J Mol Sci. 2019; 20:4574.
- [52]Chandrasekaran B, Abed SN, et al. Computer-aided prediction of pharmacokinetic (ADMET) properties. In dosage form design parameters. Academic Press: London, United Kingdom 2018, pp 731-755.
- [53]Veber DF, Johnson SR, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002; 45:2615-2623.
- [54]Yang C, Song G, et al. A review of the toxicity in fish exposed to antibiotics. Comp Biochem Physiol C Toxicol Pharmacol. 2020; 237:108840.
- [55]Ghosh AK, Panda SK, et al. Anti-vibrio and immune-enhancing activity of medicinal plants in shrimp: A comprehensive review. Fish Shellfish Immunol. 2021; 117:192-210.