Genomic identification and characterization of prophages associated with Citrobacter freundii strains
Abstract
Citrobacter freundii is a Gram-negative opportunistic bacterium that can cause infections, sepsis, and meningitis, predominantly in infants and immunocompromised adults. This study is aimed at investigating the distribution of prophages in C. freundii strains and their potential effects on the host strains using genomic characterization. In-silico analysis of 144 complete chromosomal sequences of C. freundii strains was performed, and 574 intact prophages were identified from 1178 prophages in the host. The genome sizes of the intact prophages were 6.74-115.15 kb; on average, the host chromosomes were predicted to have around 3.98% of intact prophage genomes. At least three intact prophages were most frequently predicted, while only three host chromosomes were found to have the highest number of nine intact prophages. The GC content of prophages is 50.75%, slightly lower than the average GC content (51.85%) of C. freundii. Most of the prophages in C. freundii strains were classified into four families, Myoviridae (48.16%), Siphoviridae (42%), Podoviridae (4.67%), Inoviridae (0.17%), and 5% of the intact prophages could not be assigned into any family. Phylogenomic analysis of intact prophages divided the genomes into three distinct clades. Virulence gene analysis revealed the variable distribution of 7 virulence genes (hcp, higB, hipA, msgA, rtx, yeeV, and ykfI) among the intact prophages. Overall, this study provides insights into the diversity and characteristics of prophages associated with C. freundii strains, which will help in understanding the genetic evolution and pathogenesis of the bacteria.
INTRODUCTION
Citrobacter freundii belongs to the Enterobacteriaceae family of facultative anaerobic Gram-negative bacteria [1]. They are soil-dwelling microbes in water, sewage, food, animal, and human digestive systems, sometimes responsible for community-acquired and nosocomial infections [2, 3]. C. freundii can cause a life-threatening infection that can be turned into sepsis and has also been associated with brain abscesses and newborn meningitis [4, 5]. The virulence factors of C. freundii include adhesins, endotoxin, hemolysin, and siderophores, which help it to colonize and invade host tissues [6]. Furthermore, the resistance of C. freundii to various antibiotics is also a growing concern in healthcare settings [5, 7].
Bacteriophages are viruses that infect bacteria and are abundant in the biosphere [8]. Lytic or virulent and lysogenic or temperate are the two types of bacteriophages [9]. The tailed phages are the most prominent group of viruses that affect bacteria and are the most abundant in the human gut belonging to the order Caudovirales [10]. Although the initial roles of phage genes are to infect, survive, and reproduce in the host bacteria successfully, several pathogenic and antibiotic-resistant genes are encoded by the phage genomes. Therefore, understanding the biology of the phages has several implementations in selecting phages as antibacterial agents, the evolution of bacterial strains, and typing for epidemiological studies [10]. Previously, characterization and comparative analysis of prophages present in several pathogenic strains of Burkholderia cenocepacia [11], Helicobacter pylori [12], Streptococcus mutans [13], Acinetobacter baumannii [14, 15], Acinetobacter pittii [16], Klebsiella pneumoniae [17, 18], Desulfovibrio [19], Staphylococcus aureus [20] have been performed to understand the roles of prophages in the evolution, fitness, and pathogenicity of the host bacteria.
This study elucidated the genomic characterization of 574 prophage genomes from C. freundii. Moreover, various comparative bioinformatics tools were used to study the phylogenomic, evolutionary, and virulence profile analysis of the intact prophages.
MATERIALS AND METHODS
Identification and classification of prophages sequences
The complete chromosomal sequences of 144 C. freundii strains, submitted to the National Center of Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/genome/browse/#!/prokaryotes/2850/), were retrieved until November 2022 as described in [16, 17]. Briefly, from the database C. freundii genome sequences were searched. Later, only complete C. freundii genome sequences were downloaded. Plasmid genomes were not included in the analysis. The genome size of each strain was obtained from GenBank. All the strains were screened for prophage-like elements utilizing the web server Phage Search Tool Enhanced Release (PHASTER) [21]. PHASTER was used to identify prophages as intact, questionable, or incomplete based on the length of the phage and the presence of phage-derived genes. The integrity of prophage-associated genes like integrase, endolysin, and terminase genes and the 'att' sites (attL/attR) in intact phages were manually checked for all intact prophages to reduce the likelihood of false positives. Prophage families of intact prophage were characterized by the Virfam server based on their structural compositions [22]. The recognition of head-neck-tail modules was used in Virfam for the classification of the viruses.
Prophage-associated virulence factors and antibiotic-resistance genes
Annotation of the protein sequences present in the intact prophages was performed by Rapid Annotation using System Technology (RAST) version 2.0 [23]. In Microsoft Excel, a heatmap was generated by using conditional formatting of the cells based on toxin proteins present in the intact prophages. For the prediction of antibiotic resistance genes, genomic sequences of prophages were screened by using the resistance gene identifier (RGI) tool from the comprehensive antibiotic resistance database (CARD) [24] and the ABRicate tool from Galaxy Australia [25]. The default parameters were used in all analyses conducted.
Phylogenomic analysis of intact prophage sequences
The phylogenomic study of all intact prophages was performed in Molecular Evolutionary Genetics Analysis (MEGA X) [26] using Neighbouring Joining methods with 1000 bootstrap replications. The tree was visualized by Interactive Tree of Life (iTOL) [27].
Spatial distribution of intact prophages
The spatial distribution of intact prophages inside bacterial chromosomes was performed following the protocol described in [16] using the R package ggplot2 [28]. To generate a density plot of the intact prophages, lengthwise bacterial chromosome sequences of the geom_density () function of ggplot2 were used.
RESULTS
Prevalence of prophages in the C. freundii genomes
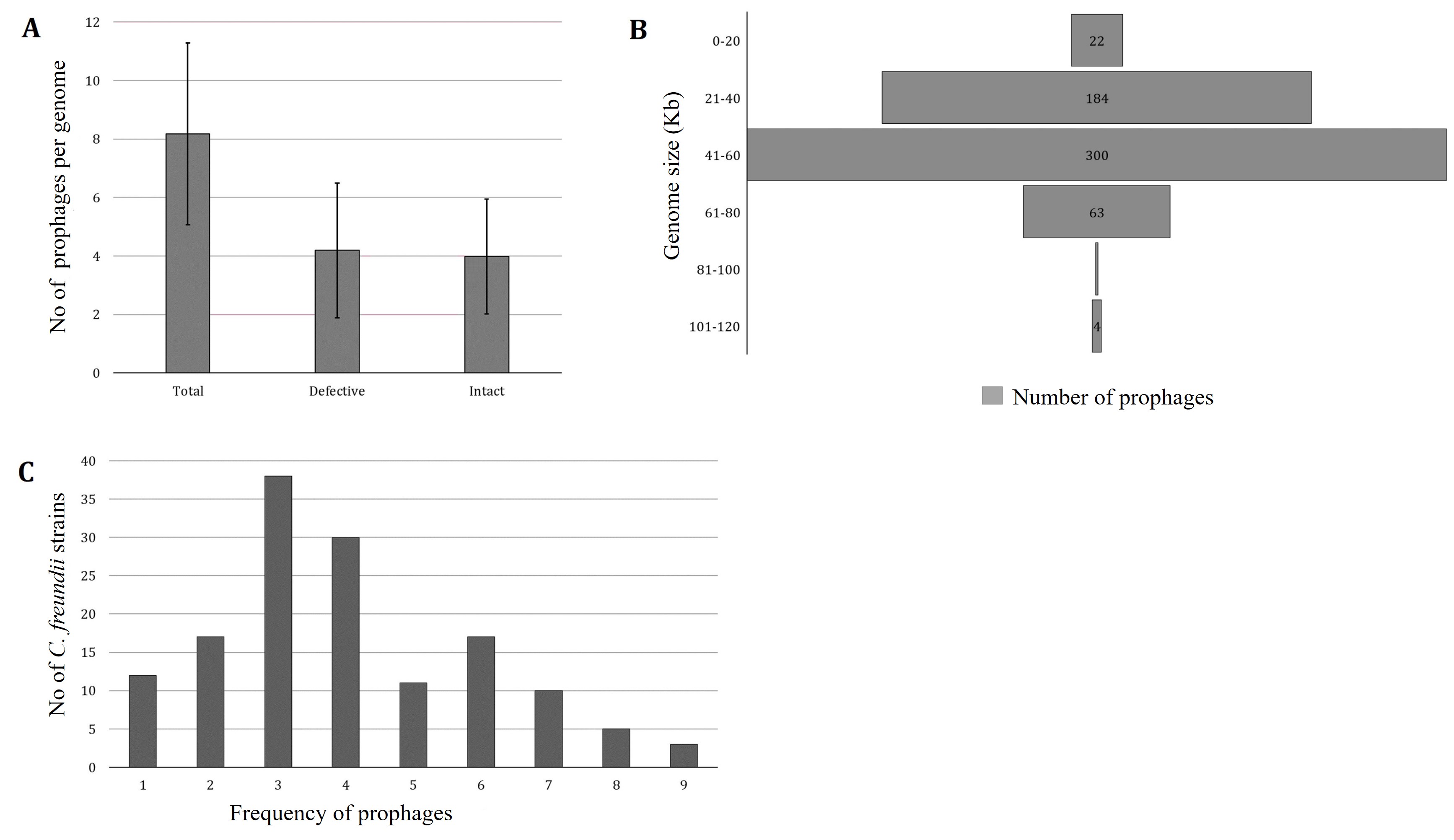
PHASTER predicted 1178 prophages from 144 C. freundii strains isolated from various sources of different countries. One strain lacks intact prophage (C. freundii CitB, Accession No: CP048416). PHASTER predicted 604 incomplete and questionable prophages and 574 intact prophages (average 3.98±1.96) (Figure 1A, Supplementary Table 1). Each chromosome, on average, had 5.2% of the total prophage genome present in C. freundii. For further analysis, we considered only intact prophages. The intact prophage sequence length ranged from 6.74 kb to 115.15 kb (Figure 1B). However, around 52% of prophages had genome sizes ranging from 41-60 kb, only one prophage ranged from 81-100 kb, and 4 prophages had genome sizes between 101-120 kb (Figure 1B). Most frequently, 3 intact prophages were identified in 38 (26.39% of 144) C. freundii. A maximum of 9 intact prophages were detected in three C. freundii (2.1%) (Figure 1C).
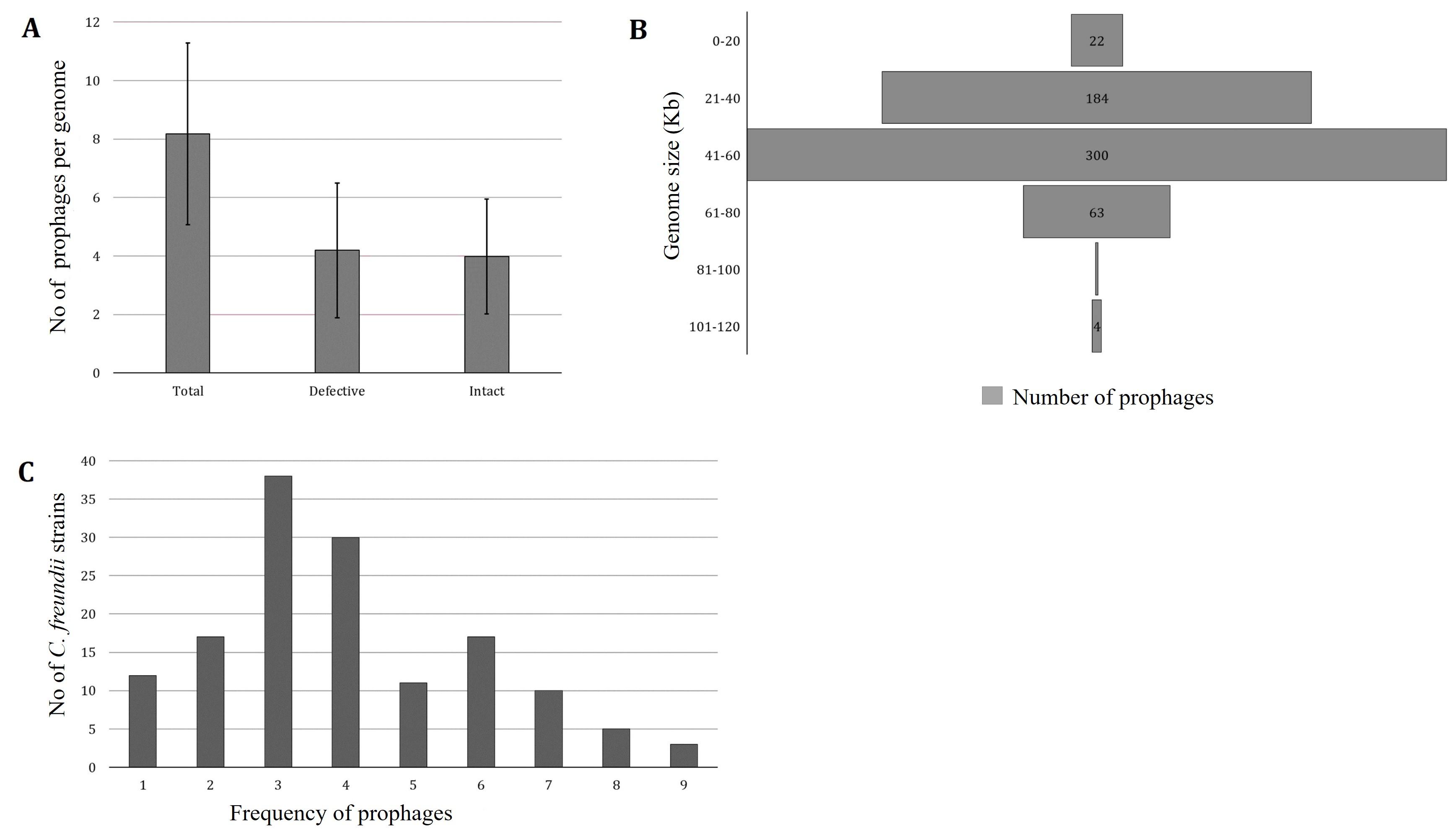
Classification of intact prophages in C. freundii strains
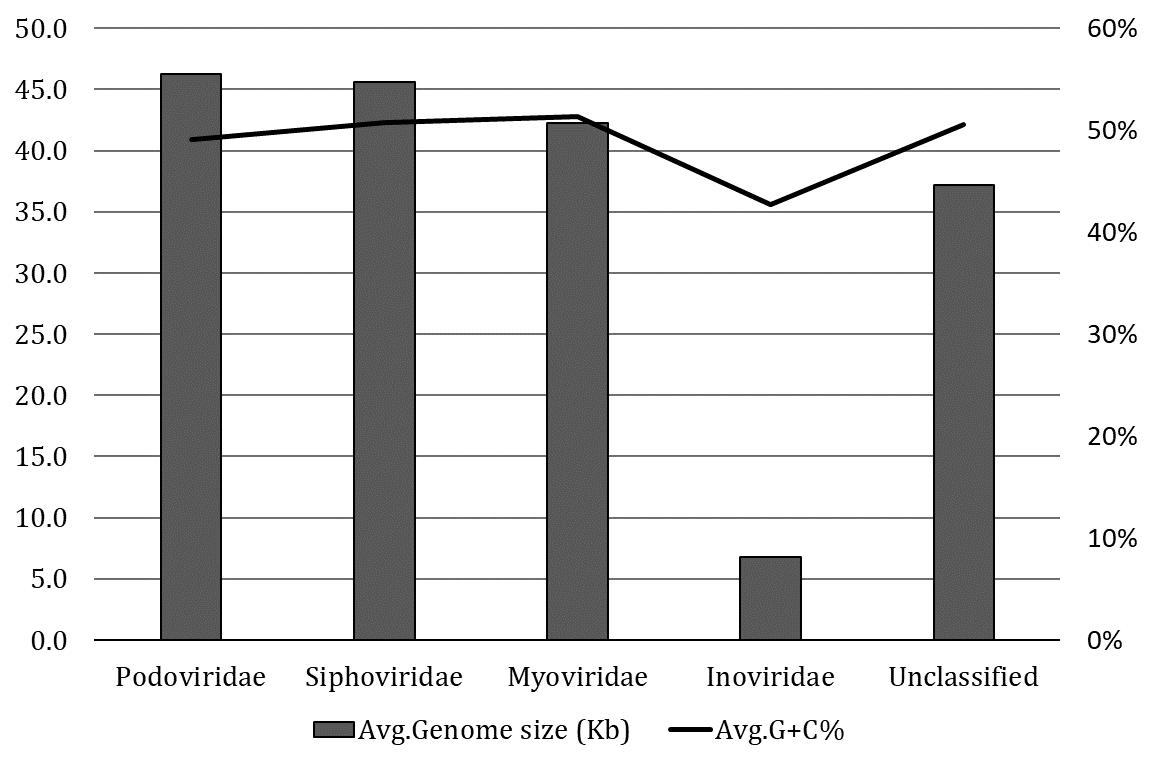
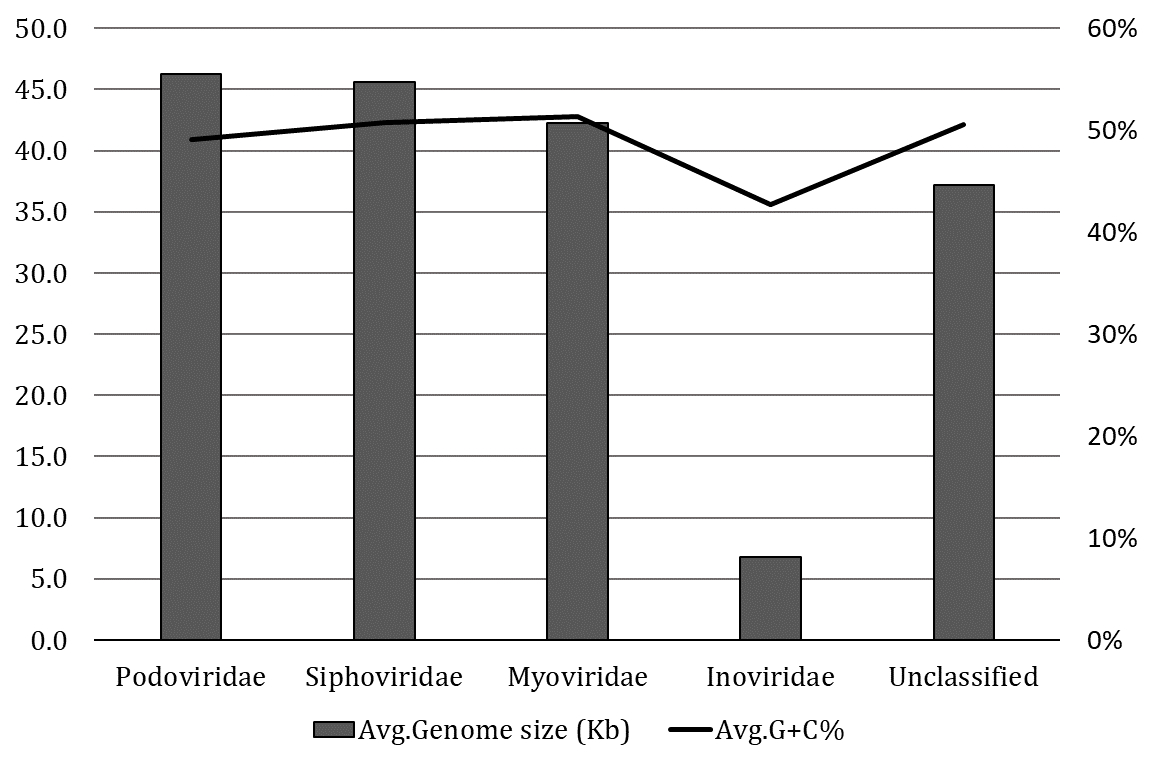
The classification of 574 intact prophages in various viral families was performed according to the proteins of head and tail connectivity, a parameter used by the VirFam tool. The majority of intact prophages (48.16%) were predicted to belong to the Myoviridae family, followed by the Siphoviridae (42%), Podoviridae (4.67%), and Inoviridae (0.17%) families. The server predicted 31(5%) unclassified prophages. The average prophage genome sizes of the Podoviridae and Siphoviridae were slightly higher than Myoviridae (Figure 2). In contrast, smaller genome sizes and lower GC contents were observed in the prophage in Inoviridae than others. This observation was due to only one prophage identified from the Inoviridae family.
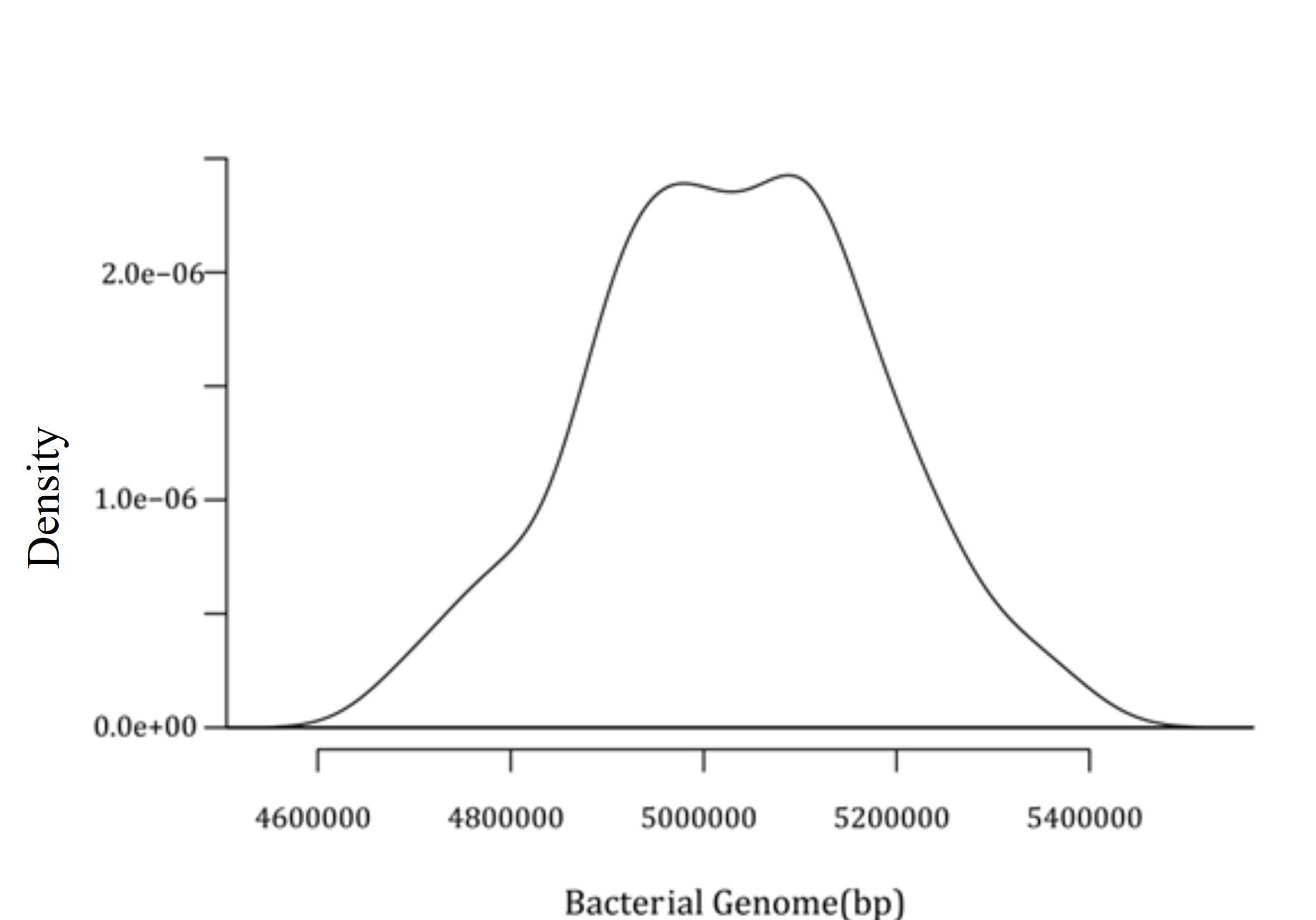
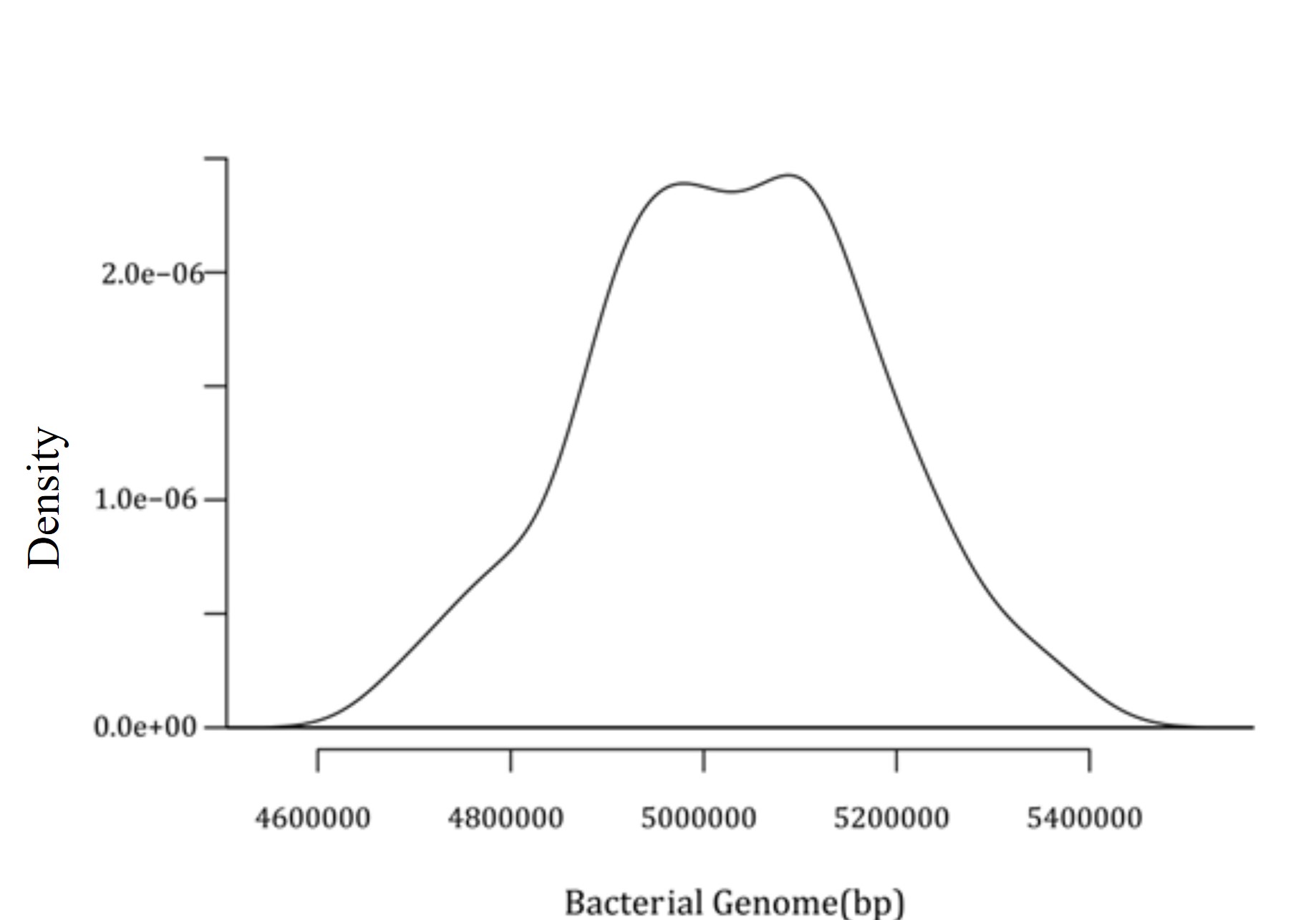
Later, a density plot for 574 intact prophages present in C. freundii chromosomes was generated. This density plot demonstrated a significant prophage density of around 54900000 bp and 5100000 bp, as shown in Figure 3.
{kind=link}
{kind=link}
{kind=link}
Presence of virulence and toxin proteins within prophages
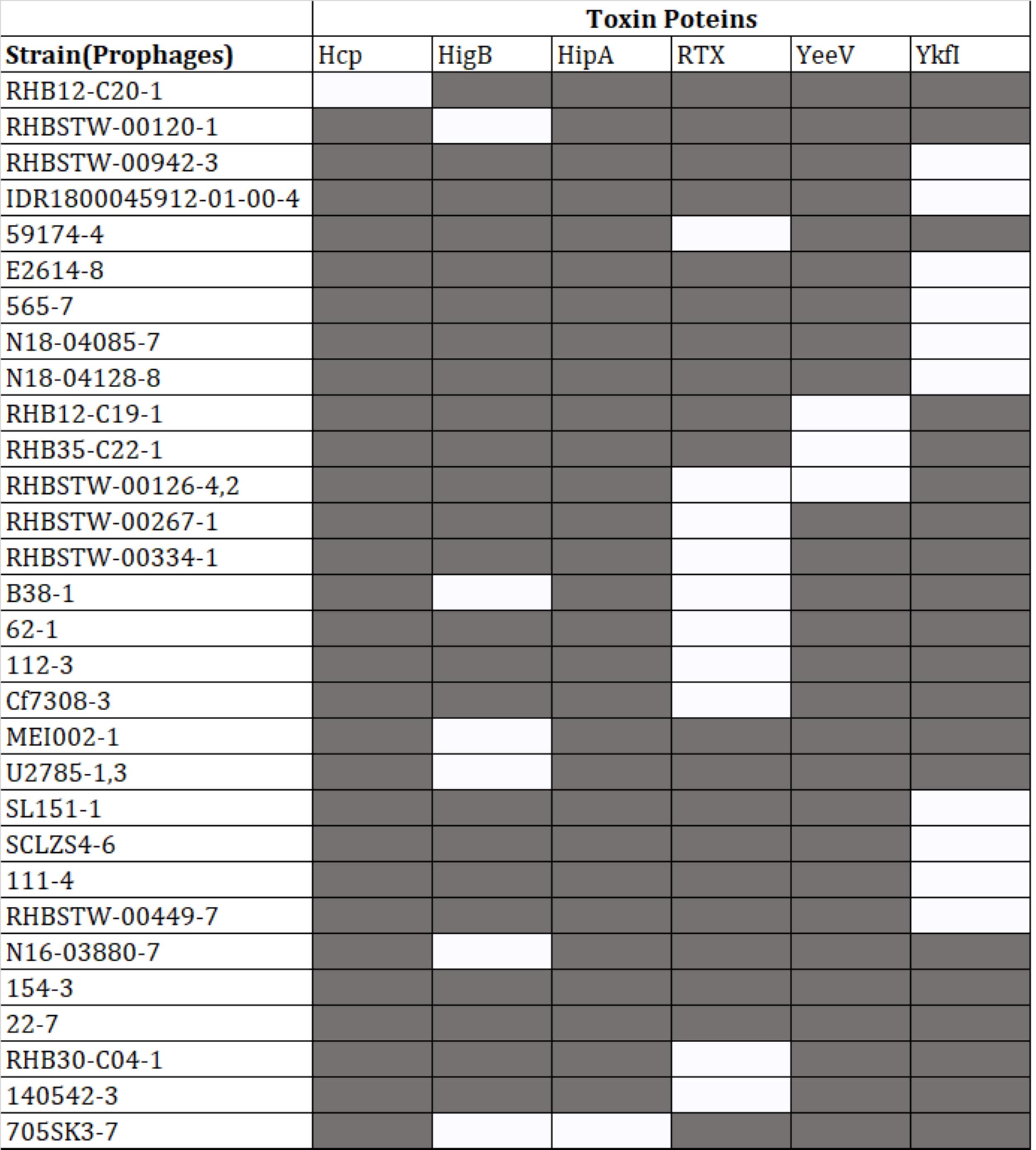
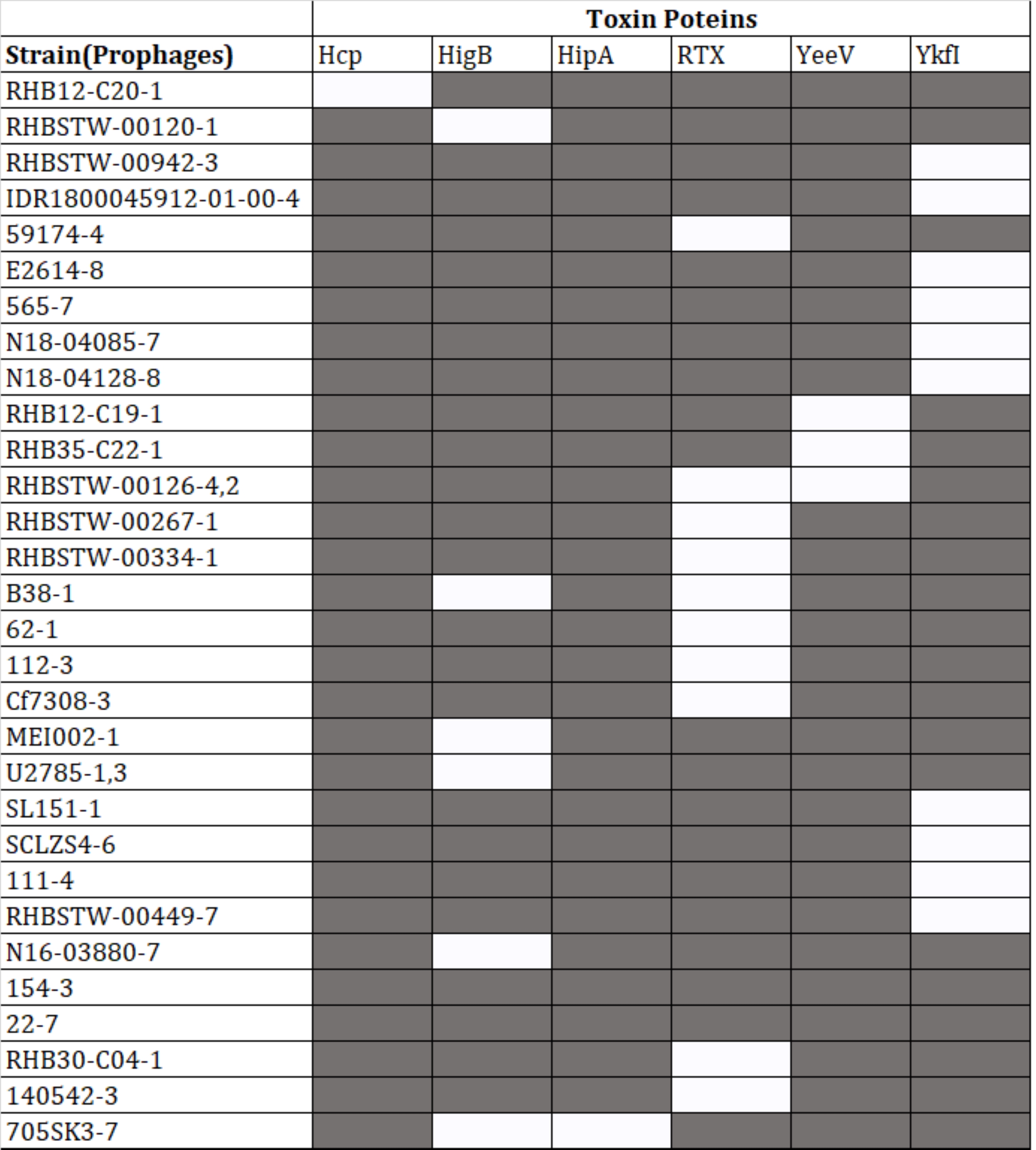
Protein-coding sequences of the intact prophages were analyzed for the presence of virulence-associated proteins. Out of 574 intact prophages, 152 (26.43%) harbored virulence genes. However, out of the 152 intact prophages, 118 contained MsgA (Macrophage-specific gunine nucleotide Exchange Factor-like) protein (Supplementary Table 2), 10 prophages were predicted to have YkfI proteins, 11 had RTX (Repeats-in-toxin) exo-proteins, 7 had HigB (one of several type III secreted effector proteins produced by E. coli, and in P. aeruginosa, HigB modulates the expression of T3SS genes and biofilm formation) proteins, 4 had YeeV proteins, and 1 prophage had Hcp (Haemolysin Co-regulated Protein) and another had HipA (high persistence A) proteins (Figure 4).
{kind=link}
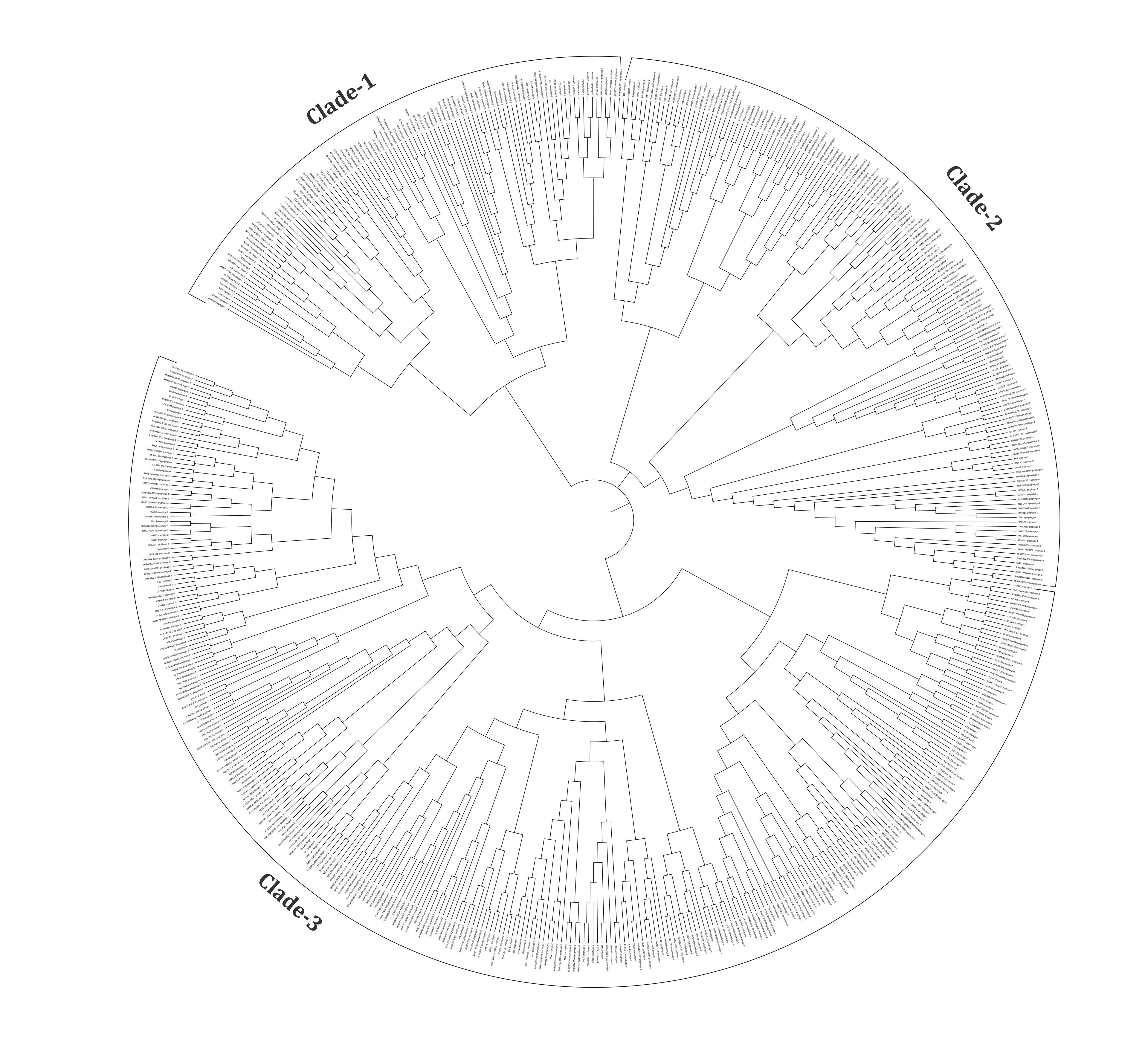
Phylogenomic analysis of the intact prophages
The whole genome sequences of 574 prophages were used to analyze the phylogenomic relationship. The results revealed that these intact prophages were classified into three major clades (Figure 5). Clade 1, clade 2, and clade 3 have 105, 155, and 314 prophage genomes, respectively. Besides, 22.86% of prophages in clade 1, 21.29% in clade 2, and 30.25% in clade 3 have virulent genes (Supplementary Table 3).
{kind=link}
DISCUSSION
Numerous bacterial isolates have prophages present, and these prophages may have contributed to the diversity of bacteria. Prophages can contribute to a bacterial strain's genetic distinctiveness by carrying different virulent components. Among other organisms, this phenomenon has been noted in Lactobacillus [29], Pseudomonas [30], Salmonella genus [10, 31], Streptococcus pyogenes [32], Streptococcus mutans [13], K. pneumoniae [17], Paenibacillus larvae [33]. Previous studies have shown that strain-specific differences are caused mainly by prophage-associated chromosomal rearrangements and deletions [17, 30, 31, 34]. Therefore, the emergence of pathogenic variants may be influenced by temperate phages carrying foreign DNA. The production of toxins and disease-modifying substances encoded by phages and phage lysogenic conversion genes can change the host phenotype and affect the virulence of the bacterial strain [35].
In this study, 1178 prophages were predicted in 144 C. freundii strains, with an average of 5.2% of the host chromosome content prophage genome, which is lesser than other bacterial genomes (10-20%) [35, 36]. On the other hand, a more significant fraction of prophages in C. freundii were found to be defective (51.27%). The incomplete and questionable prophages frequently lack essential phage functions. Such deficiency might be caused by the selective pressure of the host to inactivate mutations on the newly acquired phages. Consequently, the causative genes of infection were deleted or mutated, providing multiple benefits to the host bacterium to adjust to hostile environmental conditions [37]. Therefore, our further analysis focused on intact prophages characterized by an integrase domain, transcriptional regulator, or at least one of the structural domains attributed to either capsid, tailed, or portal proteins. The prophage GC content is 50.75%, lower than the average GC content of 51.85% in the C. freundii chromosomes. The intact prophage sizes in C. freundii range from 6.1 kb to 399.9 kb. In contrast, another member of the Enterobacteriaceae family had the prophage sizes in K. pneumoniae range from 29.7 kb to 50.6 kb [18], 6.4 kb to 358 kb for Salmonella [31], and 8.1 kb to 31.9 kb for Shigella [38]. There seems to be a massive difference in the prophage sizes of the members of the Enterobacteriaceae family. Similar prophages in different host isolates indicate their horizontal movement events and significance in genomic plasticity or evolution (Supplementary Table 4) [39]. C. freundii strains were shown to have multiple phages ranging from 10 to 17, belonging to different families and clades in different groups. Multiple prophage-like elements suggest that poly-lysogeny may be shared in many bacterial pathogens, including C. freundii.
The viral families are the basic unit of the biological classification of viruses, and these are characterized mainly based on physiochemical properties, genome structures, morphology, and gene expression [22]. Among the 574 intact prophages studied, the In silico approaches revealed Myoviridae as the most represented family (48.16%), followed by Siphoviridae (42%), Podoviridae (4.67%) and Inoviridae (0.17%). Similar to our studies, another investigation focusing on the prophages of K. pneumoniae revealed that 62 prophages in Myoviridae (59.6%), followed by 40 prophages in Siphoviridae (38.5%) and, 2 prophages in Podoviridae (1.9%) could be classified [18]. Furthermore, the prophages in A. baumannii could be divided into Myoviridae (25.69%), Siphoviridae (31.20%), Podoviridae (10.09%), and unclassified (33.02%) [40]. In C. freundii, Myoviridae is typically the most prominent phage family and has below-average genome sizes (42.25 kb). In comparison, Siphoviridae (45.62 kb) and Podoviridae (46.25 kb) had sizes slightly above the average (42.97 kb) (Figure 2). A phylogenomic tree was generated and grouped into three major clades to understand the identified diversity of intact prophages (Figure 5, Supplementary Table 3). All the clades were revealed to be a mixed sub-cluster with higher diversity, comprising prophages from Myoviridae, Siphoviridae, Podoviridae, Inoviridae, and unclassified families based on the program used in the study.
Among the 574 intact prophages, the most dominating are Klebsiella phage phiKO2 (9.93%) and Salmonella phage 118970_sal3 (9.58%) (Supplementary Table 4). These two phages are present in several C. freundii strains isolated from several locations. The 574 intact prophages studied showed that some shared more than 95% of their genome identities, indicating solid evolutionary relationships. We discovered 65 phages that shared more than 70% similarities with the 574 intact prophages regarding identity when compared to the studied prophage genomes against NCBI databases (Supplementary Table 4).
Multiple studies have shown the relationship between the pathogen's virulence and the acquisition of prophages leading to numerous infectious diseases. Some prophages carry genes that can change the characteristics of the host, ranging from increased host fitness to virulence [41-43]. Even defective prophages are thought to be capable of transporting virulence factors [44]. In our 152-virulence gene containing intact prophages analysis, msgA was predominantly distributed. (Supplementary Table 2). The MsgA exhibits a distinctive expression pattern during macrophage infection that contributes to yeast cell morphogenesis variation [45]. Additionally, MsgA has been suggested to play a role in the biofilm formation and antibiotic resistance in Bacillus subtilis and S. aureus [46, 47]. Therefore, this protein may contribute to biofilm formation and antibiotic resistance in C. freundii.
Some C. freundii prophages have been found to have T1SS-secreted agglutinin RTX protein with diverse biological functions like virulence, biofilm formation, and resistance to antibiotics [48]. But in Citrobacter spp. the activities of T1SS-secreted agglutinin RTX proteins are still unknown, although the prevalence of RTX protein was the second highest (~6.57%) after the MsgA virulence factor identified in the current study. A previous study showed that RTX-containing proteins often have hemolytic or hemagglutinating activity in B. cenocepacia J2315 and identified four hemolysin-type Ca2+-binding domains at the carboxy-terminal of the protein sequence that is characteristic of RTX-related proteins [49]. Therefore, some prophages confer new virulence factors and/or antibiotic resistance genes used for bacterial pathogenesis and beneficial traits like increased fitness to the host [44, 50].
Furthermore, we have found a gene for the Hcp family protein in one of the intact prophages (Figure 4). The Hcp family of proteins is secreted through the Type 6 Secretion System (T6SS), which plays a crucial role in the pathogenesis of bacterial infections and contributes to the virulence of the invading bacteria, including E. coli, Vibrio, Aeromonas, and Pseudomonas [51-53]. According to previous research, the T6SS genomic island in C. freundii CF74 strain modulates the flagellar system, improves motility, thus is involved in adhesion to host cells, and produces cytotoxicity in host cells [54]. This study detected the toxin gene hipA in one of the intact prophages from C. freundii strain 705SK3. HipA toxin is a part of the HipA-HipB Type II Toxin Antitoxin (TA) system, where HipA is involved in the inhibition of Glutamyl-tRNA synthetase, thereby ceasing protein synthesis and leading to the death of the bacteria [53, 55]. HipA-HipB Type II, TA system, was reported in the IncA/C1 Plasmid pCf587 of C. freundii 33587 [56].
The heat map analysis revealed the presence of another three different proteins, Ykfl, HigB, and YeeV, among the remaining intact prophages associated with virulence factors. One study conducted using E. coli YeeV toxin reported that YeeV interacts with two major cytoskeleton proteins, FtsZ (a tubulin-like protein) and MerB (a prokaryotic actin-like protein), to inhibit the activities of both GTPase and GTP-dependent polymerization of FtsZ as well as the ATP-dependent polymerization of MerB. Consequently, it inhibits cell division and changes the morphology and lysis of E. coli [57]. Another study reported that prolonged overproduction of YkfI causes complete cell death within seconds or minutes by interacting with the cell division protein FtsZ in cryptic prophages of E. coli K-12 [58]. It is also evident from the same study that toxin YkfI constitutes a toxin-antitoxin (TA) pair with type IV antitoxin YafW. Such antitoxin can only partially block the toxicity of YkfI. However, the interaction of YkfI with FtsZ did not cause cell death in Pseudomonas strains [58]. Several studies reported the role of toxin HigB, a member of the ribosome-dependent RelE toxin family, in association with Type II antitoxin HigA in regulating pathogenicity in many opportunistic pathogenic bacteria, including E.coli K-12, P. aeruginosa PA14, Vibrio cholerae, Streptococcus pneumoniae [55, 59, 60]. Therefore, further research is needed to determine whether C. freundii prophages can benefit the host's evolutionary fitness due to these virulence factors.
CONCLUSION
To summarize, this study successfully identified the intact prophages in all completely sequenced strains of C. freundii that were previously unknown. This study also investigated the genomes of these prophages to determine their potential impact on the host strains. C. freundii, an opportunistic pathogen, harbors prophages that could affect its virulence and cause genetic rearrangements. Nearly half of the identified prophages were incomplete, suggesting that C. freundii is actively integrating and modifying its prophages. The intact prophages may actively influence the bacterial genome and contribute to the pathogenicity of C. freundii by encoding significant virulence genes. Many prophages may be crucial in bacterial evolution, adaptability, and physiology. Furthermore, conducting prophage induction experiments would provide insights into host-phage interactions. The research findings also indicate the presence of multiple prophage-like elements, known as poly-lysogeny, which can convert non-pathogenic bacteria into pathogenic ones capable of producing harmful toxins. Additionally, the results demonstrate that C. freundii and other members of the Enterobacteriaceae family share similar patterns in terms of prophage distribution and the prevalence of common prophages.
ACKNOWLEDGEMENT
Authors would like to acknowledge the Department of Biochemistry and Microbiology, North South University for their technical support for the project.
AUTHORS CONTRIBUTION
Conceptualization: SRS; Data curation: SIM, ABL, AA, SHO; Formal analysis: SIM, AR, ABL, IJ; Methodology: SRS, ABL, SIM, IJ; Writing – original draft: IJ, SIM, SI, SRS; and Writing – review & editing: SRS, IJ, SI. All authors have read and approved the final version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
SUPPLEMENTARY MATERIALS
Supplementary Table 1. Characterization of the prophage genomes in C. freundii genomes, Supplementary Table 2. List of Toxin and virulence proteins, Supplementary Table 3. Distribution of the prophages in different clades, and Supplementary Table 4. Representative prophages present in the genomes of C. freundii (Supplementary Materials).
References
- [1]Manouore Njoya A, Poutoum Yogne Y, et al. Antibiotic susceptibility of four enterobacteriaceae strains (enterobacter cloacae, Citrobacter freundii, Salmonella typhi and Shigella sonnei) isolated from wastewater, surface water and groundwater in the equatorial zone of cameroon (central africa). World Journal of Advanced Research and Reviews. 2021;11:120-37.
- [2]Liu L, Chen D, et al. Genetic diversity, multidrug resistance, and virulence of Citrobacter freundii from diarrheal patients and healthy individuals. Front Cell Infect Microbiol. 2018;8:233.
- [3]Rostamzad A, Karami Cherag Abad A, et al. Antibiotic resistance of Citrobacter freundii in clinical isolates: A systematic review and meta-analysis. Journal title. 2019;6:58-63.
- [4]Chen D, Ji Y. New insights into Citrobacter freundii sepsis in neonates. Pediatr Int. 2019;61:375-80.
- [5]Liu LH, Wang NY, et al. Citrobacter freundii bacteremia: Risk factors of mortality and prevalence of resistance genes. J Microbiol Immunol Infect. 2018;51:565-72.
- [6]Hossain S, Wimalasena SHMP, et al. Virulence factors and antimicrobial resistance pattern of Citrobacter freundii isolated from healthy pet turtles and their environment. Asian Journal of Animal and Veterinary Advances. 2017:10-6.
- [7]Rahman A, Shamsuzzaman SM, et al. Antimicrobial susceptibility pattern and virulence genes detection in Citrobacter freundii isolated from patients of a tertiary care hospital, Bangladesh. The International Arabic Journal of Antimicrobial Agents. 2022;12:1-7.
- [8]Sharma S, Chatterjee S, et al. Bacteriophages and its applications: An overview. Folia Microbiologica: Springer Netherlands; 2017. p. 17-55.
- [9]Hartley MA, Ronet C, et al. Backseat drivers: The hidden influence of microbial viruses on disease. Current Opinion in Microbiology. 2012. p. 538-45.
- [10]Gao R, Naushad S, et al. Comparative genomic analysis of 142 bacteriophages infecting Salmonella enterica subsp. enterica. BMC Genomics. 2020;21:374.
- [11]Roszniowski B, McClean S, et al. Burkholderia cenocepacia prophages—prevalence, chromosome location and major genes involved. Viruses. 2018;10.
- [12]Lu W, Wise MJ, et al. Comparative analysis of the full genome of Helicobacter pylori isolate sahul64 identifies genes of high divergence. Journal of Bacteriology. 2014;196:1073-83.
- [13]Fu T, Fan X, et al. Comparative analysis of prophages in Streptococcus mutans genomes. PeerJ. 2017;2017.
- [14]Loh B, Chen J, et al. A biological inventory of prophages in Acinetobacter baumannii genomes reveal distinct distributions in classes, length, and genomic positions. Frontiers in Microbiology. 2020;11:1-13.
- [15]Turner D, Ackermann HW, et al. Comparative analysis of 37 Acinetobacter bacteriophages. Viruses. 2018;10:1-25.
- [16]Zhu R, Mathur V. Prophages present in Acinetobacter pittii influence bacterial virulence, antibiotic resistance, and genomic rearrangements. PHAGE: Therapy, Applications, and Research. 2022;3:38-49.
- [17]Baliga P, Shekar M, et al. Genome-wide identification and analysis of chromosomally integrated putative prophages associated with clinical Klebsiella pneumoniae strains. Current Microbiology. 2021;78:2015-24.
- [18]Marques AT, Tanoeiro L, et al. Genomic analysis of prophages from Klebsiella pneumoniae clinical isolates. Microorganisms. 2021;9.
- [19]Crispim JS, Dias RS, et al. Screening and characterization of prophages in Desulfovibrio genomes. Scientific Reports. 2018;8:1-10.
- [20]Naorem RS, Goswami G, et al. Comparative analysis of prophages carried by human and animal-associated Staphylococcus aureus strains spreading across the european regions. Scientific Reports. 2021;11:1-13.
- [21]Arndt D, Grant JR, et al. PHASTER: A better, faster version of the phast phage search tool. Nucleic Acids Research. 2016;44:W16--W21.
- [22]Lopes A, Tavares P, et al. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics. 2014;15.
- [23]Aziz RK, Bartels D, et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:1-15.
- [24]Alcock BP, Raphenya AR, et al. Card 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:D517-d25.
- [25]Jalili V, Afgan E, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Research. 2021;48:W395--W402.
- [26]Kumar S, Stecher G, et al. MEGAX: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547-9.
- [27]Letunic I, Bork P. Interactive tree of life (iToL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Research. 2021;49:W293-W6.
- [28]Wickham H. Elegant graphics for data analysis: Ggplot2. Applied Spatial Data Analysis with R. 2008:21-54.
- [29]Figueroa-Bossi N, Uzzau S, et al. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Molecular Microbiology. 2001;39:260-72.
- [30]Ha AD, Denver DR. Comparative genomic analysis of 130 bacteriophages infecting bacteria in the genus Pseudomonas. Frontiers in Immunology. 2018;9:1-13.
- [31]Fong K, Lu YT, et al. Prophage diversity across Salmonella and verotoxin-producing Escherichia coli in agricultural niches of British Columbia, Canada. Frontiers in Microbiology. 2022;13:1-12.
- [32]Nakagawa I, Kurokawa K, et al. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Research. 2003;13:1042-55.
- [33]Ribeiro HG, Nilsson A, et al. Analysis of intact prophages in genomes of Paenibacillus larvae: An important pathogen for bees. Frontiers in Microbiology. 2022;13.
- [34]Ventura M, Canchaya C, et al. Integration and distribution of Lactobacillus johnsonii prophages. Journal of Bacteriology. 2003;185:4603-8.
- [35]Ronning CM, Losada L, et al. Genetic and phenotypic diversity in Burkholderia: Contributions by prophage and phage-like elements. BMC Microbiology. 2010;10.
- [36]Kim SH, Park JH. Characterization of prophages in Leuconostoc derived from kimchi and genomic analysis of the induced prophage in Leuconostoc lactis. Journal of Microbiology and Biotechnology. 2022;32:333-40.
- [37]de Sousa JAM, Buffet A, et al. Modular prophage interactions driven by capsule serotype select for capsule loss under phage predation. ISME Journal. 2020;14:2980-96.
- [38]Sváb D, Falgenhauer L, et al. Genome analysis of a historical Shigella dysenteriae serotype 1 strain carrying a conserved stx prophage region. Frontiers in Microbiology. 2021;11:1-8.
- [39]Kopotsa K, Mbelle NM, et al. Epigenomics, genomics, resistome, mobilome, virulome and evolutionary phylogenomics of carbapenem-resistant Klebsiella pneumoniae clinical strains. Microbial Genomics. 2020;6:1-19.
- [40]Costa AR, Monteiro R, et al. Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness. Scientific Reports. 2018;8:1-12.
- [41]Boyd EF. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Advances in Virus Research: Academic Press Inc.; 2012. p. 91-118.
- [42]Fortier LC, Sekulovic O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence. 2013;4:354-65.
- [43]Hayashi T, Makino K, et al. Complete genome sequence of enterohemorrhagic Eschelichia coli O157:H7 and genomic comparison with a laboratory strain k-12. DNA Research. 2001;8:11-22.
- [44]Wang X, Kim Y, et al. Cryptic prophages help bacteria cope with adverse environments. Nature Communications. 2010;1.
- [45]Weerasinghe H, Bugeja HE, et al. The novel Dbl homology/BAR domain protein, MsgA, of talaromyces marneffei regulates yeast morphogenesis during growth inside host cells. Scientific Reports. 2021;11:1-19.
- [46]Branda SS, González-Pastor JE, et al. Genes involved in formation of structured multicellular communities by Bacillus subtilis. Journal of Bacteriology. 2004;186:3970-9
- [47]Lindsay JA. Staphylococcus aureus genomics and the impact of horizontal gene transfer. International Journal of Medical Microbiology. 2014;304:103-9.
- [48]Linhartová I, Bumba L, et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiology Reviews 2010. p. 1076-112.
- [49]Whitby PW, VanWagoner TM, et al. Identification of an RTX determinant of Burkholderia cenocepacia J2315 by subtractive hybridization. Journal of Medical Microbiology. 2006;55:11-21.
- [50]Bobay L-MM, Touchon M, et al. Pervasive domestication of defective prophages by bacteria. Proceedings of the National Academy of Sciences. 2014;111:12127-32.
- [51]Hood RD, Singh P, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host and Microbe. 2010;7:25-37.
- [52]Suarez G, Khajanchi BK, et al. Actin cross-linking domain of Aeromonas hydrophila repeat in toxin a (rtxa) induces host cell rounding and apoptosis. Gene. 2012;506:369-76.
- [53]Wen Y, Behiels E, et al. Toxin-antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathogens and Disease. 2014;70:240-9.
- [54]Liu L, Hao S, et al. The type VI secretion system modulates flagellar gene expression and secretion in Citrobacter freundii and contributes to adhesion and cytotoxicity to host cells. Infection and Immunity. 2015;83:2596-604.
- [55]Wood TL, Wood TK. The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiology Open. 2016;5:499-511.
- [56]Ruggiero M, Girlich D, et al. Complete sequence of the inca/c1 plasmid pcf587 carrying blaper-2 from Citrobacter freundii. Antimicrob Agents Chemother. 2018;62:1-6.
- [57]Tan Q, Awano N, et al. Yeev is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol Microbiol. 2011;79:109-18.
- [58]Wen Z, Wang P, et al. Interaction of type IV toxin/antitoxin systems in cryptic prophages of Escherichia coli k-12. Toxins. 2017;9:1-14.
- [59]Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966-76.
- [60]Xu BS, Liu M, et al. Conformational changes of antitoxin higa from Escherichia coli str. K-12 upon binding of its cognate toxin HigB reveal a new regulation mechanism in toxin-antitoxin systems. Biochem Biophys Res Commun. 2019;514:37-43.