Effect of growth factors present in serum on insulin resistance and endothelial dysfunction in EA.hy926 cells
Abstract
Insulin resistance is a pathophysiological function of Type II diabetes mellitus which can be comprehended by quantifying the parameters critical to the insulin signaling pathway. Serum has a profound role in evaluating cellular growth and metabolism in vitro. We hypothesize that the growth factors present in serum such as IGF, EGF, and FGF have an effect on the regulatory components of the insulin signaling pathway leading to insulin resistance. This study focuses on the metabolic effect of Fetal Bovine Serum (FBS) in endothelial (EA.hy926) cells. A dose and time-dependent treatment of FBS on the chosen cells was followed by assessing cell viability and glucose uptake capacity using MTT and 2-NBDG assays respectively. Spectrophotometric analysis of nitric oxide (NO) and lactate dehydrogenase (LDH) determined vascular homeostasis and no cytotoxic effects of the serum, respectively, in endothelial cells. These findings indicate that FBS at higher levels could possibly lead to loss of NO activity which in turn could impair endothelium-mediated dilation. The inhibition of the enzymatic activity of eNOS, which in response to the stress may activate the release of LDH in endothelial cells displaying cytotoxicity. In conclusion, our findings indicate that a specific concentration of serum exposure enhances insulin signaling and endothelium cell regulation by modulating glucose uptake and NO production.
INTRODUCTION
Type Ⅱ diabetes mellitus (T2DM) is a syndrome caused by a combination of metabolic impairments such as insulin receptor (IR) action, insulin receptor substrate (IRS), and phosphatidylinositol 3 kinase (PI3K) signaling. These aspects individually or in amalgamation with the others are devoted to the deterioration of vascular function and possibly lead to diabetic complications such as loss of endothelial cell function. A broad group of secreted polypeptides that are relatively small and stable are known as the growth factors [1]. They play a significant role in cell growth, proliferation, differentiation, and cell signaling. Through the activation of specific transmembrane receptors in cells, relaying of the stimuli of the growth factor into the cell occurs [2]. Serum is a powerhouse of several growth factors utilized in cell proliferation, differentiation, and growth. Fetal bovine serum (FBS) is inarguably used as an essential growth supplement in animal cell culture media and consists of several growth factors – EGF, FGF, VEGF, and IGF [3]. We aim to investigate the effect of these metabolic factors on insulin signaling cascades. Even though prior studies have established the signaling effect of growth factors in causing insulin resistance, the metabolic aspect of the factors present in the serum along with the media is yet to be explored. Insulin is one among the factors which upon stimulation causes the phosphorylation of the insulin receptor substrate thereby leading to the activation of various downstream signaling aspects [4]. Insulin also is responsible for the dilation of blood vessels and improves the overall flow of blood throughout the body. These blood vessels consist of a group of cells, including endothelial cells (EC) and vascular smooth muscle cells (VSMC) which coordinate and regulate vaso homeostasis [5]. Endothelial cells have a vasomotor function in blood vessels, so the coordination of EC and VSMC maintains and regulates vaso homeostasis. For this process, Endothelial nitric oxide synthase (eNOS) acts as a marker of endothelial cell functions and produces NO, which undergoes paracrine signaling with associated cells in blood vessels. eNOS is the third isoform of the nitric oxide synthase gene (NOS3) and is one of the two constitutive isoforms of the gene [6]. Nitric Oxide (NO) (a vasodilator in the endothelium) development is induced by Insulin which brings about a boost in blood circulation and decreased glucose uptake [7]. At a cellular level, the PI3K-dependent insulin signaling pathway regulates endothelial-NO production by a potential enzymatic activity of endothelial Nitric oxide synthase (eNOS) [8]. L-arginine is converted to NO by the eNOS expression. Additionally, it results in the regulation of vasoconstrictor endothelin-1 (ET-1) by the MAPK-dependent insulin signaling pathway [9].
This study focuses on serum (FBS), which in a concentration-dependent manner may ameliorate insulin resistance and develop a compensatory mechanism to enhance the signaling aspects of insulin-stimulated events such as glucose uptake, vaso homeostasis and metabolism in EA.hy926 endothelial cells. The specific aims were achieved by performing a time course study following cell viability and cytotoxicity assessment. Also, the metabolic effect on cells was determined by observing the glucose uptake capacity and levels of nitric oxide.
MATERIALS AND METHODS
Reagents and chemicals
Insulin, Fetal Bovine Serum (FBS), and Dulbecco’s Modified Eagle Medium (DMEM), LDH assay kit were procured from HiMedia, India. Other reagents such as MTT (Thiazolyl Blue Tetrazolium Bromide), cell culture tested from HiMedia, Nω-nitro-l-arginine methyl ester (L-NAME) > 98% TLC, powder from Sigma-Aldrich, (N-(1-Naphthyl) ethylenediamine dihydrochloride, Hi-AR/ACS) (NED) from HiMedia, and Sulfanilamide, Hi-ARTM > 99% from HiMedia were purchased.
Cell culture of EA.hy926 cells
EA.hy926 cells (passage no. 2) were obtained from HiMedia, India and were maintained at 37℃ in a CO2 incubator. The cells were grown in DMEM with 10% Fetal Bovine Serum. The EA.hy926 were procured at passage number 2 and it takes 2 -3 days to reach 75% confluency. EA.hy926 cells, after reaching 75% confluence in three days were used for the entire study.
Cell viability test
Following a 2-hour period of serum starvation, the viability of the cells with respect to varying serum concentrations (5%,10%,15% for 5min, 15min and 30min each) was analyzed by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay. Triton X-100 was used as the positive control and DMSO was used as the solubilization buffer to dissolve the purple colour formazan product. Percentage cell viability was calculated after measuring the absorbance at 570nm [10].
Nitric oxide estimation
The nitric oxide test for endothelial cells was performed with the help of the Griess assay. The supernatant from the cell viability test was stored and used. A red-pink color develops after the addition of the NED (N-(1-Naphthyl) ethylenediamine) and sulfanilamide solutions. The intensity of this colour was measured at an absorbance of 540nm. The percentage of nitric oxide was calculated using the OD and slope values [11].
Cytotoxicity estimation by the release of lactate dehydrogenase
The cell cytotoxicity of EA.hy926 cells were estimated with the help of LDH assay with varying serum (FBS) concentrations (5%, 10%, 15%) and varying time intervals (5min, 15min, 30min). Triton X-100 (0.01%) served as the positive control and the control (untreated cells) served as negative control. The initial solution-A and stop solution that was added in the dark to the supernatant were used from the LDH assay kit, after which the absorbance was measured at 580nm. Percentage cytotoxicity was then calculated from the OD values obtained [12].
Measurement of 2-NBDG glucose uptake
The glucose uptake for endothelial cells was measured using a 2-NBDG (D-Glucose, 2-deoxy-2-((7-nitro-2,1,3-benzoxadiazol-4-yl) amino)-glucose) assay, with 2-NBDG being a glucose analog. Insulin was used as a positive control. The glucose uptake was measured with respect to the varying serum concentrations (5%, 10%, 15%) with varying time intervals. Excitation wavelengths of 485 or 355nm and emission wavelengths of 538 or 460nm were used to test fluorescence intensity [13].
Statistical analysis
Mean ± SEM were represented. The statistical significance was evaluated using one-way ANOVA in GraphPad Prism 8.0.2 to determine differences among groups. The criterion for statistical significance was *p < 0.05 and #p < 0.001.
RESULTS
Effect of serum treatment on cell viability
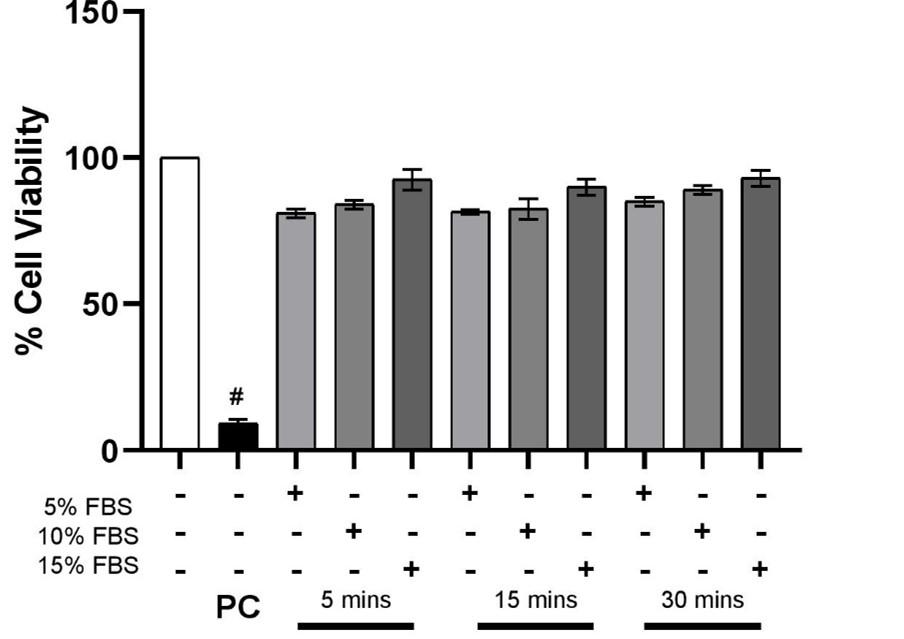
As described above, our initial aim was to determine the effect of exposure time and concentration of the FBS on the cells. The viability assay performed using MTT established that 15% FBS displayed maximum live cell count at each time point – 5 min, 15 mins and 30 mins. However, as the difference between cell viability among the concentrations and time points remained insignificant (p>0.05), it was concluded that the serum did not have a time and dose-dependent effect on the viability of EA. hy926 cells in comparison to the positive control, Triton-X 100 (Figure 1).
Effect of serum treatment on nitric oxide levels
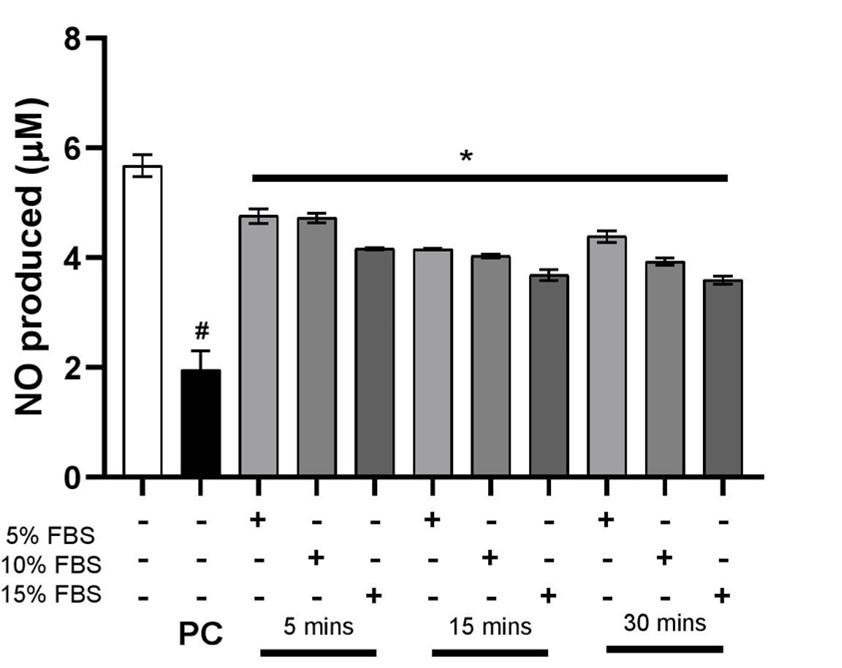
The endothelial cells following a dose and time-dependent FBS treatment were subjected to eNOS inhibitor L-NAME to compare the production of nitric oxide by untreated cells. The graph in Figure 2 shows that the production of NO is significantly reduced in the cells treated with L-NAME whereas the levels in basal cells remained unchanged.
Effect of serum treatment on cell cytotoxicity
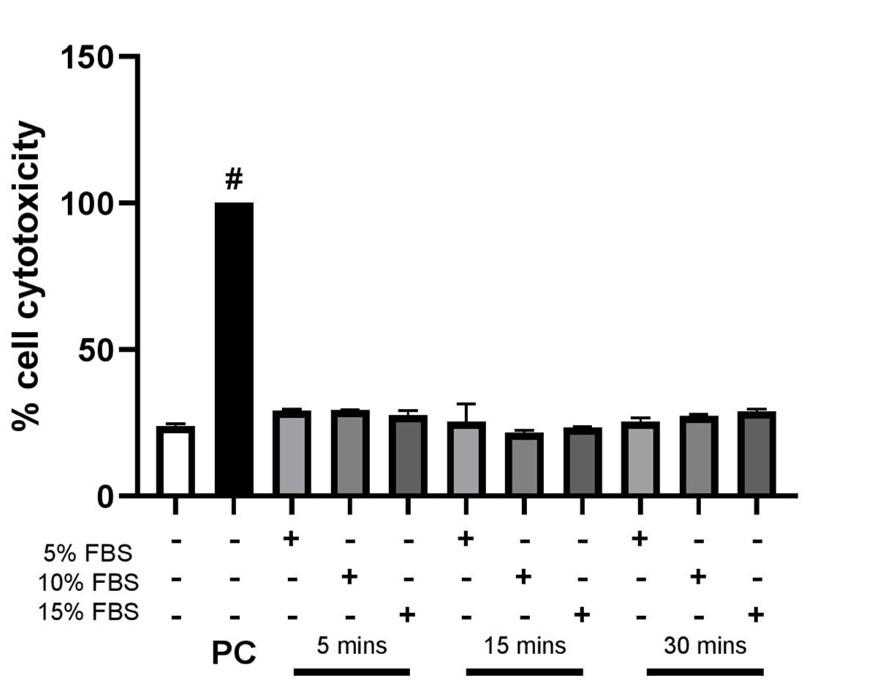
The endothelial cells were treated with FBS in a dose and time-dependent manner. The amount of LDH produced is increased when cells are exposed to Triton X-100 which is used as a positive control. It can be observed from the graph in Figure 3 that all the values are similar to the control and hence it can be concluded that FBS does not have any significant cytotoxic effect on the endothelial cells.
Effect of serum treatment on 2-NBDG glucose uptake
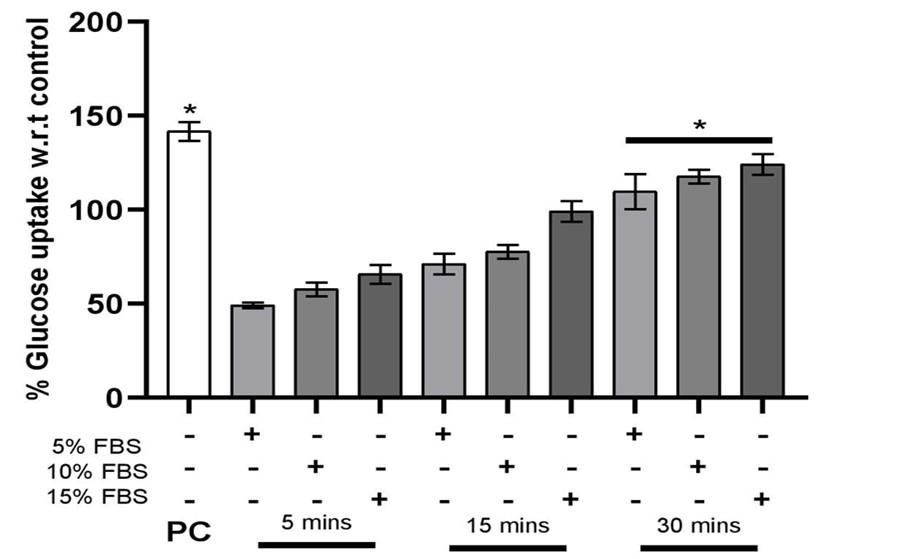
The endothelial cells were exposed to FBS in a time and dose-dependent method. To ascertain the intracellular glucose accretion, a fluorescent glucose analogue (2-NBDG) was utilized to observe the uptake in individual cells [12]. In Figure 4, in the case of positive control due to insulin, we observe higher uptake of glucose in the cells.
The percentage of glucose uptake by endothelial cells gradually increases, which were treated with serum, suggesting that as the concentration and incubation time increases a significant increase is observed in the uptake of glucose by the cells.
DISCUSSION
In this study we provide conclusive biological evidence that treatment of endothelial cells with serum at three serial concentrations trigger GLUT-4 activation followed by glucose uptake and eNOS enzymatic activity for endothelial cells function [14]. We demonstrated that serum does not impact cell viability, whereas increased concentrations of serum was found to be non-cytotoxic to EA. hy926 cells.
With insulin-induced eNOS phosphorylation and the reduction of IRS-2 expression, impaired insulin signaling in the endothelial cells reduces insulin-induced glucose uptake by the skeletal muscle via, at least in part, decreased capillary recruitment and decreased interstitial insulin concentrations in the skeletal muscle. As it happens, insulin signaling defect induced by IRS-2 deletion from the endothelial cells simultaneously impedes insulin-induced glucose uptake by the skeletal muscle, escalation in concentration of interstitial insulin and debilitation of the insulin-induced capillary recruitment. This insight into the mechanism also sheds light on the physiological roles of IRS-2 in the endothelial cells. Normal glucose homeostasis crucially seems to accommodate expression of a sufficient amount of IRS-2 in the endothelial cells. While there is surplus in IRS-2 expression during the fasting state, induction of sufficient glucose uptake by the skeletal muscle takes place and after feeding the raised glucose levels return to within the normal range. However, during the fasting period when endothelial cell expression of IRS-2 is decreased in the presence of hyperinsulinemia with insulin resistance, impairment in insulin signaling occurs, and the elevated levels of glucose after feeding fail to reduce efficaciously. Our results demonstrates that a fluorometric assay that is used to determine the glucose taken up by the cells in-turn aids in analyzing the cell metabolism as well as signaling characteristics. Thereby it is a mechanistic study of obesity and diabetes [15]. The percentage of glucose absorption by serum-treated endothelial cells, suggesting that as the concentration and incubation time increases a significant increase is observed in the uptake of glucose by the cells. This can be explained by the improved expression of the glucose transporters such as GLUT-4. Insulin stimulates the mobilization of vesicles containing GLUT-4 glucose transporters to the plasma membrane, resulting in glucose transfer into the cells. In case of positive control due to hyperinsulinemia, caused by insulin dysfunction, higher uptake of glucose is observed in the cells. As a result, we may infer that a higher serum concentration aids in the reduction of insulin resistance by improving glucose uptake activity.
Generally, both vasorelaxation and vasoconstriction are induced by insulin. Mediation of insulin-induced vasorelaxation is carried out by the IRS-PI3K-AKT pathway elevating endothelial NO production, whereas Shc/SOS/Ras-MAPK pathway inducing ET-1 expression mediates insulin-induced vasoconstriction [16]. One of the principal features of the pathophysiology of diabetes is oxidative stress. Glycolysis metabolite methylglyoxal (MG) was formerly observed to affect redox balance by stimulating ROS production and modulating the expression and function of cytoprotective molecules such as glutathione [17], superoxide dismutase [18], glyoxalase [19] and H2S [20]. Increased NADPH oxidase-derived O2•− reacts with eNOS-derived NO in order to create ONOO− that in turn oxidizes BH4 to BH2. In variety of conditions such as hyperphenylalaninemia, pathogenesis implies decrease in BH4 [21]. Improvement was observed in diabetes [22], ischemia reperfusion injury [23], hypertension [24], Alzheimer disease and Parkinson’s disease [25], and in many of these pathological conditions due to exogenous BH4 [21,23] or sepiapterin [26,27, 28, 29] supplementation. BH4 is key in eNOS dimers maintenance and furthermore is functionally related to S-glutathionylation, a powerful eNOS uncoupling regulator [29]. Our study demonstrates that Nitric oxide is considered one of the main regulators of energy metabolism and body composition and hence, measuring the amount of nitric oxide will help determine the condition and function of the cells [30]. Although no significant conclusions can be derived based on the results of the cells treated with serum, a possible hypothesis could be made which explains that increased concentrations of serum might decrease the development of Nitric Oxide. Studies have shown that a drop in NO production from L-arginine in endothelial cells is a major factor for insulin resistance in diabetic patients [31,32]. This can be explained by the idea that the cocktail of growth factors contained in the serum such as EGF, FGF and VEGF may have an adverse effect on the eNOS enzymatic activity leading to the depletion in NO production when compared to control. Endothelial dysfunction resulting in a decreased level of NO activity can not only guide vasoconstriction but can also impair endogenous negative feedback loop resulting in the inhibition of expression of certain pro-inflammatory cytokines [33]. This decreased bioavailability of NO could be responsible for endothelial dysfunction which can be confirmed by performing further gene expression studies of certain specific targets [8].
FBS serves as an exogenous source of LDH, which may have a significant consequence for cytotoxicity analysis [30]. LDH, whose major role is to guide cellular respiration, is also associated with endothelial-derived soluble adhesion molecules and recognizes markers of endothelial activation that are commonly suppressed by NO. LDH is let into the bloodstream or into other bodily fluids when damage to tissues, organs, or the cells occurs. Similarly, when the cells are exposed to serum at three different concentrations for three different time intervals, in any case of the damage to the cells, there occurs a possibility for LDH to be released. Over expression of LDH may often interfere with normal glucose metabolism and insulin secretion by beta cells, and thus may be directly responsible for insulin secretory defects in some diabetes mellitus II cases [34]. Testing the amount of LDH in the supernatant will establish the extent of cytotoxic effect of the serum on cells [35]. From the results obtained, we can conclude that the amount of LDH produced is maximum when the endothelial cells are exposed to Triton X-100 which is used as a positive control. Among all the cells treated at different concentrations for different time periods, the ones treated for 30 minutes with a serum concentration of 15% exhibited higher LDH levels. However, these levels are not sufficient to be considered cytotoxic to the cells and hence, do not show proof of the serum possibly causing an insulin resistant condition due its high concentrations.
A cross talk study provides evidence stating that the insulin-mediated AKT and eNOS activations are induced optimally in the endothelial cells after feeding, resulting in insulin-induced capillary recruitment, interstitial insulin concentration increase, as well as elevation of skeletal muscle glucose. Due to inadequacy in the insulin-mediated AKT and eNOS activations in the endothelial cells of obese subjects after feeding, the insulin-induced capillary recruitment, interstitial insulin concentration increases and raise of skeletal muscle glucose uptake are damaged. With this information our study emphasized that serum does not have an impact on GLUT-4 translocation followed by glucose uptake, as understood at the cellular level of eNOS function on endothelial cells. However, the importance of insulin in balancing metabolic activity and endothelial cells functions and its crosstalk is remains unexplored at molecular level.
CONCLUSION
This study focuses on observing if serum (FBS) might ameliorate insulin resistance and develop a compensatory mechanism to enhance the signaling aspects of insulin-stimulated events such as glucose uptake, vaso homeostasis, and metabolism in EA.hy926 endothelial cells, when given in a concentration-dependent and time-dependent manner.
After the treatment with serum increasing concentrations and the respective time periods, it was observed that not only does serum have no cytotoxic effect on the cells but also improves their viability. This determination can be supported by the significant increase in glucose uptake by the cells with an increase in serum concentrations. The maintenance of natural glucose homeostasis in the body depends on GLUT-4-mediated glucose transport in the cells [36]. As an increased amount of serum is provided for a prolonged time, the cells tend to make use of the high glucose environment which improves GLUT-4 expression and thereby reduces the insulin resistant condition which can support the increased glucose tolerance due to high GLUT-4 expression [37]. LDH also associated with endothelial-derived soluble adhesion molecules, recognized markers of endothelial activation that are commonly suppressed by NO [38]. From the results of the LDH assay, it can be understood that even though the cells produce higher amounts of LDH at increasing concentrations, their concentrations are not significant enough to establish that serum has an ameliorating effect on insulin resistance. The reduction in NO could be attributed to the condition of endothelial dysfunction thereby hindering vaso homeostasis. The conclusion drawn should further be confirmed with the help of gene expression studies that are done by targeting and understanding the specific factors involved like eNOS, and GLUT-4 to establish an appropriate hypothesis of the insulin resistance mediated endothelial dysfunction (Figure 5).
Figure 5. Effect of growth factors in serum on insulin resistance and endothelial dysfunction. In the ideal pathway, IRS is phosphorylated by Insulin, which activates the MAP Kinase signaling pathway for cell growth, proliferation and gene expression, as well as the PI3K signaling pathway for GLUT-4 translocation, lipid synthesis and cell proliferation. Insulin resistance results in the disruption of these signaling pathways. ET-1 is a vasoconstrictor, while NO is a vasodilator. Endothelial dysfunction occurs as a result of IR disrupting the balance between these levels by decreasing the NO levels and increasing the ET-1 levels.
ACKNOWLEDGEMENT
None.
AUTHORS CONTRIBUTION
HM, PT and AP drafted the manuscript. HM, PT, RK, AP and AB performed the experiments. AM proofread the manuscript and helped in analyzing the data. SS designed the experiments, analyzed, corrected, and validated the results. All authors read and approved the final submitted version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Behm B, Babilas P, Landthaler M, Schreml S. Cytokines, chemokines and growth factors in wound healing. Journal of the European Academy of Dermatology and Venereology 2012;26:812–20.
- [2]Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 2017;9:1–45..
- [3]Subbiahanadar Chelladurai K, Selvan Christyraj JD, Rajagopalan K, Yesudhason BV, Venkatachalam S, Mohan M, et al. Alternative to FBS in animal cell culture – An overview and future perspective. Heliyon 2021;7:e07686.
- [4]Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harbor perspectives in biology 2014;6: a009191.
- [5]Barac A, Campia U, Panza JA. Methods for evaluating endothelial function in humans. Hypertension 2007;49:748–60.
- [6]Król M, Kepinska M. Human nitric oxide synthase—its functions, polymorphisms, and inhibitors in the context of inflammation, diabetes and cardiovascular diseases. International Journal of Molecular Sciences 2021;22:1–18.
- [7]Sandoo A, Veldhuijzen van Zanten JJCS, Metsios GS, Carroll D, Kitas GD. The Endothelium and Its Role in Regulating Vascular Tone. The Open Cardiovascular Medicine Journal 2015;4:302–12.
- [8]Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Reviews in Endocrine and Metabolic Disorders 2013;14:5–12.
- [9]Meza CA, la Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? International Journal of Molecular Sciences 2019;20.
- [10]Davis JA, Sharma S, Mittra S, Sujatha S, Kanaujia A, Shukla G, et al. Antihyperglycemic effect of Annona squamosa hexane extract in type 2 diabetes animal model: PTP1B inhibition, a possible mechanism of action. Indian Journal of Pharmacology 2012;44:326–32.
- [11]Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radical Biology and Medicine 2007;43:645–57.
- [12]Thomas MG, Marwood RM, Parsons AE, Parsons RB. The effect of foetal bovine serum supplementation upon the lactate dehydrogenase cytotoxicity assay: Important considerations for in vitro toxicity analysis. Toxicology in Vitro 2015;30:300–8.
- [13]Blodgett AB, Kothinti RK, Kamyshko I, Petering DH, Kumar S, Tabatabai NM. A fluorescence method for measurement of glucose transport in kidney cells. Diabetes Technology and Therapeutics 2011;13:743–51. https://doi.org/10.1089/dia.2011.0041.
- [14]Su Y, Qadri SM, Wu L, Liu L. Methylglyoxal modulates endothelial nitric oxide synthase-associated functions in EA.hy926 endothelial cells. Cardiovascular Diabetology 2013;12:134.
- [15]Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. Journal of Biochemical and Biophysical Methods 2005;64:207–15.
- [16]Muniyappa R, Quon MJ. Is further research needed on glucosamine?. The Journal of family practice 2007;56:343.
- [17]Price CL, Knight SC. Methylglyoxal: possible link between hyperglycaemia and immune suppression? Trends in Endocrinology and Metabolism 2009;20:312–7.
- [18]Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. The Journal of Biological Chemistry 2003;278:1728–34.
- [19]di Loreto S, Zimmitti V, Sebastiani P, Cervelli C, Falone S, Amicarelli F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. International Journal of Biochemistry and Cell Biology 2008;40:245–57.
- [20]Chang T, Untereiner A, Liu J, Wu L. Interaction of methylglyoxal and hydrogen sulfide in rat vascular smooth muscle cells. Antioxidants & redox signaling 2010;12:1093–1100.
- [21]Kim HK, Ha SH, Han J. Potential therapeutic applications of tetrahydrobiopterin: From inherited hyperphenylalaninemia to mitochondrial diseases. Annals of the New York Academy of Sciences 2010;1201:177–82.
- [22]Abudukadier A, Fujita Y, Obara A, Ohashi A, Fukushima T, Sato Y, et al. Tetrahydrobiopterin Has a Glucose-Lowering Effect by Suppressing Hepatic Gluconeogenesis in an Endothelial Nitric Oxide Synthase-Dependent Manner in Diabetic Mice. Diabetes 2013;62:3033–43.
- [23]Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Katano T, et al. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. Journal of molecular and cellular cardiology 2011;50:534–44.
- [24]Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, et al. Pivotal Role for Endothelial Tetrahydrobiopterin in Pulmonary Hypertension. Circulation 2005;111:2126-33.
- [25]Foxton RH, Land JM, Simon AE, Heales JR. Tetrahydrobiopterin Availability in Parkinson’s and Alzheimer’s Disease; Potential Pathogenic Mechanisms. Neurochemical research 2007;32:751–756.
- [26]Cheng H, Wang H, Fan X, Paueksakon P, Harris RC. Improvement of endothelial nitric oxide synthase activity retards the progression of diabetic nephropathy in db/db mice. Kidney International 2012;82:1176–83.
- [27]Thida M, Earl J, Zhao Y, Wang H, Tse CS, Vickers JJ, et al. Effects of Sepiapterin Supplementation and NOS Inhibition on Glucocorticoid-Induced Hypertension. American journal of hypertension 2010;23:569–74.
- [28]Tiefenbacher CP, Lee CH, Kapitza J, Dietz V, Niroomand F. Sepiapterin reduces postischemic injury in the rat heart. European journal of physiology 2003;447:1–7.
- [29]Crabtree MJ, Brixey R, Batchelor H, Hale AB, Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. Journal of Biological Chemistry 2013;288:561–9.
- [30]Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Current Opinion in Clinical Nutrition and Metabolic Care 2010;13:97–104.
- [31]Wu G, Meininger CJ. Nitric oxide and vascular insulin resistance. BioFactors 2009;35:21–7.
- [32]Arnal JF, Münzel T, Venema RC, James NL, Bai CL, Mitch WE, et al. Interactions between L-arginine and L-glutamine change endothelial NO production: An effect independent of NO synthase substrate availability. Journal of Clinical Investigation 1995;95:2565–72.
- [33]Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vascular health and risk management 2005;1:183–98.
- [34]Ainscow EK, Zhao C, Rutter GA. Acute overexpression of lactate dehydrogenase-A perturbs β-cell mitochondrial metasbolism and insulin secretion. Diabetes 2000;49:1149–55.
- [35]Kaja S, Payne AJ, Naumchuk Y, Koulen P. Quantification of lactate dehydrogenase for cell viability testing using cell lines and primary cultured astrocytes. Current Protocols in Toxicology 2017;72:1–10.
- [36]Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nature Medicine 2000;6:924–28.
- [37]Wang T, Wang J, Hu X, Huang XJ, Chen GX. Current understanding of glucose transporter 4 expression and functional mechanisms. World Journal of Biological Chemistry 2020;11:76–98.
- [38]Kato GJ, McGowan V, Machado RF, Little JA, Taylor VI J, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 2006;107:2279–85.