Intermittent androgen therapy in prostate cancer reveals the pro-apoptotic roles of androgen/androgen receptor: an overview
Abstract
For the treatment of advanced prostate cancer, either castration alone or in conjunction with androgen ablation is a crucial therapeutic strategy. Patients initially respond favorably to the treatment but eventually reach a hormone-resistant stage known as an androgen refractory tumor, which has an aggressive propensity to spread. Changes in the control of apoptotic pathways have been linked to this development towards androgen unresponsiveness. Reduction in apoptosis sensitivity or elevation in resistance to it seems to be a significant indicator of oncogenic transformation that cannot be treated. Resuming androgen levels after intermittent androgen therapy has been proposed to change tumor cells' growth behavior and make them more sensitive to pro-apoptotic drugs. This review offers an overview of the current understanding of the therapeutic benefits of androgen/androgen receptor-induced apoptotic induction. It also sheds light on the implications of activating novel apoptotic pathways in prostate cancer cells with regard to resistance targeting.
INTRODUCTION
With its high death rates, prostate cancer is among the most serious illnesses worldwide [1,2]. Millions of men are impacted by this disease each year, making it the second cause of cancer-related mortality in the United States, with an annual incidence of 300,000 cases [3]. The majority of prostate malignancies are treated with androgen deprivation therapy (ADT) as a cornerstone [4]. But after a short while, the tumor often returns in a more aggressive form, leading to the development of androgen-independent prostate cancer (AIPC) [5]. A change in the apoptotic pathways has been suggested as the most likely mechanism for the advancement of AIPC [6]. Thus, it would be interesting to investigate and comprehend how androgen/androgen receptor (A/AR) signaling cascades induce apoptotic pathways in prostate malignancies.
The regulation of gene expression is facilitated by the ligand-activated transcription factor known as the androgen receptor (AR) [7]. AR is composed of various domains both structurally and functionally. The C-terminus's ligand-binding domain (LBD) is where androgen binding occurs. The N-terminal domain (NTD) is where the transactivation activity of AR mainly takes place, and the DNA-binding domain (DBD) is where functional dimerization and recognition of Androgen Response Elements (AREs) take place [8-11]. AR is prevented from binding DNA and rendered inactive in the cytosol prior to ligand binding by its interaction with heat shock proteins. The androgen receptor undergoes a transformation upon binding its ligand, releasing it from the heat shock proteins and allowing it to be translocated into the nucleus. There, it binds DNA as a homodimer on AREs, forming a variety of complexes with the basal transcription apparatus to control the expression of different genes involved in the growth, differentiation, and apoptosis of prostate cells [12,13].
Homeostasis is developed and maintained in large part by the process of controlled cell death known as apoptosis. Genetic and epigenetic changes are linked to cancer cells' ability to evade apoptosis [14]. One of the key stages in the development of tumors is abnormalities in the apoptotic signaling pathways [15]. It is clear that compromised apoptosis increases prostate cancer cells' resistance to many types of treatment [16]. During intermittent androgen therapy (IAT), alterations in the growth behavior of tumor cells are linked to the restoration of androgen levels [17]. Early on, intermittent androgen therapy increases the propensity of cancer cells to undergo apoptosis and preserves some degree of differentiation through regeneration [18,19]. These altered behaviors may increase their androgen reliance, which could postpone the onset of hormone-independent cancer while minimizing damage and enhancing response to treatment drugs [18, 20, 21].
Reduced expression of AR led to increased kinase activity and Ca2+/calmodulin-dependent protein kinases II (CaMkII) gene expression. This caused the PI3k/Akt pathway to become activated, which in turn caused apoptosis to be evaded [19]. It has been proposed that androgen deprivation eliminates their growth-inhibiting function [22]. Research has demonstrated that giving androgen to castrated mice has the ability to change androgen-independent prostate cancer into an androgen-stimulated form [23]. In order to replace malfunctioning apoptotic pathways and treat prostate cancer, androgen therapy can be viewed as a therapeutic strategy. This could slow the disease's progression and incidence [24]. The data reported in this review indicate that medicines that enhance apoptosis may be able to further enhance the apoptotic tendency of tumor cells, which may be the case when using IAT instead of ADT in the treatment of prostate cancer [25–27].
METHODS
The material was gathered using the keywords "androgen on prostate cancer" and "androgen on apoptosis" from published online research databases (1979–2023), including PubMed and Google Scholar. The Microsoft PowerPoint was used to generate the figures.
APOPTOTIC ROLE OF A/AR IN PROSTATE CANCER CELL MODELS
Different levels of AR expression, ranging from low (PC-3(AR)13) to high (PC-3(AR)10), were observed in a variety of PC-3 clonal cell lines transfected with a cytomegalovirus (CMV) promoter [28]. It revealed a relationship between the androgenic effect on growth inhibition and the expression level of AR [29]. AR negative cell lines, PC-3(AR) cells [30,31], and DU145 [32,33] were similarly shown to have an apoptotic response upon stable transfection with AR in the presence of androgens [30,34]. LNCaP-derived sublines, including MOP [19], R2 [32], 104-R [33], ARCaP [35], and LNCaP-LNO cells [36], demonstrated decreased cell cycle progression with an elevated apoptotic index when treated with androgens [22]. These results demonstrated the relationship between AR expression and growth inhibition in various cell lines.
The differentiation effects of androgen to cause apoptosis were proven by stable transfected AR in the non-tumorigenic cell line HPr-1 [37]. When treated with androgens, it demonstrated growth arrest between the G1 and S phases with a more differentiated morphology in the HPr-1AR cell line as compared to HPr-1[37]. Nevertheless, the androgenic growth suppressive action was totally eliminated by pre-treating these cells with anti-androgens such as hydroxyflutamide (HF), indicating a potential communication between the apoptotic signaling cascade and AR/A [37]. Therefore, evaluating the molecular and cellular interactions of A/AR in different PCa cell lines (Table 1) is crucial in order to have a better knowledge of apoptotic induction in prostate cancer. This will undoubtedly hold great promise in the creation of efficient therapeutic approaches.
Table 1. The role of A/AR in apoptosis of prostate cancer in cellular models.
ANDROGEN/AR-MEDIATED MOLECULAR TARGETING OF APOPTOSIS
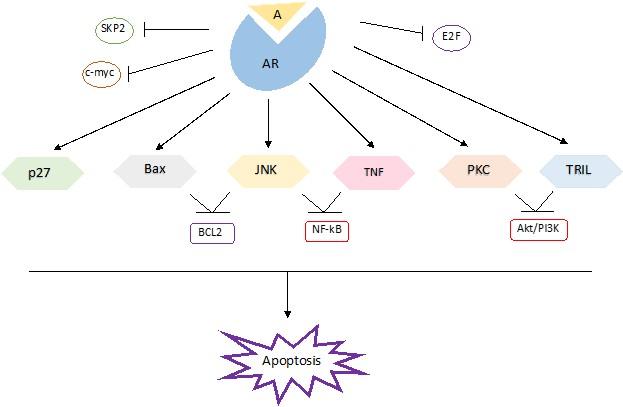
This section summarized various mechanisms of apoptosis, including the direct mechanism of A/AR induces apoptosis (Figure 1) and the indirect mechanism of A/AR induces apoptosis (Figure 2).
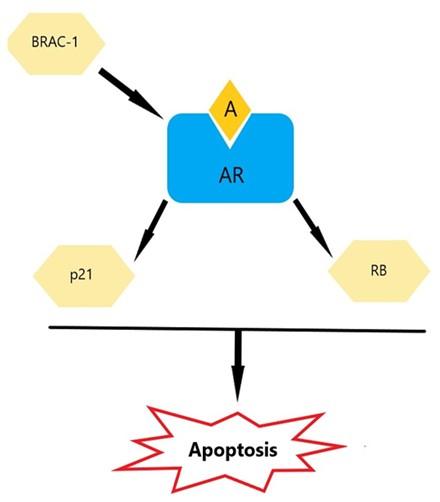
Retinoblastoma protein
Retinoblastoma protein (RB) controls the course of the cell cycle to prevent tumor growth through processes including apoptosis and differentiation [43]. RB overexpression in LNCaP cells has been shown to be linked to apoptosis [44]. It was discovered that tumors lacking in RB alone did not grow as well in castrated mice with decreased RB levels [45], suggesting a potential relationship between RB and AR. RB's activation of AR requires the ligand, even though binding has been shown to be ligand-independent [46]. The synergistic functional relationship between AR and RB to trigger apoptosis in prostate cancer is largely dependent on the transcriptional activity of AR and the intact pocket domains of RB [40]. A considerable increase in AR transcriptional activity was seen in a study using co-transfected Rb and AR in DU-145 cells (lacking both AR and RB). This was followed by the development of a mitochondrial-dependent apoptotic pathway when androgen was added to the medium [40, 46]. The phosphorylation and dephosphorylation states of RB may be a mechanism tying the functional connection between RB and A/AR [46]. Rb can only suppress tumors when it is in its active, hypophosphorylated form [47]. Through the upregulation of CDKIs, androgens are known to activate RB by inhibiting its phosphorylation at serine 780 (Ser780) and serine 795 (Ser795) [48]. Consequently, a poor treatment response is associated with the disruption of Rb function that is seen during androgen deprivation [49]. These findings highlight the significance of the synergy between RB and AR in tumor suppression
Bcl-2–associated X protein
A proapoptotic member of the Bcl-2 family, Bcl-2-associated X protein (Bax), induces apoptosis via an intrinsic mechanism [50]. Bax sensitizes the cells to numerous stimuli that induce cell death in addition to its role as a tumor suppressor in prostate cancer [51, 52]. In order to cause cell death, the androgen receptor stimulates the translocation of Bax and its interaction with its targets in the outer mitochondrial membrane in a ligand-dependent manner [53]. Bax sensitizes the cells to numerous stimuli that induce cell death in addition to its role as a tumor suppressor in prostate cancer [52, 54]. To cause cell death, the androgen receptor stimulates the translocation of Bax and its interactions with additional molecules in the outer membrane of the mitochondria in a ligand-dependent manner [53]. By creating heterodimers with Bax, Bcl-2 blocks the intrinsic pore-forming activity of Bax, which may be the source of its pro-apoptotic actions [55]. Therefore, in prostate cancer, the Bcl-2 to Bax ratio plays a significant role in regulating the sensitivity to Bax-induced cell death [56]. It has been demonstrated that androgens can change this ratio in Bax's favor by lowering Bcl-2 levels. The Bcl-2 pathway is covered in greater detail later in this article. Despite the androgen absence, AR can cause Bax-mediated apoptosis via a non-genotropic effect, but androgen is required to make the cells more sensitive to Bax's apoptotic effects [53, 57, 58]. This impact was mediated by the transcriptional activity of AR triggered by androgens, which was eliminated when AR expression in AR-positive LNCaP 104-R1, 104-S, and CDXR cells was suppressed by employing AR siRNA [59]. Simultaneously, the resistant PC-3 cells were rendered susceptible to Bax-induced death by transfection of AR expression [57-60].
p27 (Cip/Kip 1)
Additionally, known as cyclin-dependent kinase inhibitor 1B (Cip/Kip 1), p27 functions as a tumor suppressor by inducing differentiation and inhibiting the cell cycle [61]. A reduction in the level of p27 expression was seen in most cases of advanced prostate cancer and androgen-refractory prostate cancer [39, 62]. Studies have shown a correlation between the elevation of p27 expression in these cells and the androgenic reduction of 104-R1 and 104-R2 proliferation [63]. Androgens have been shown to produce a long-lasting increase in p27 levels, even at low dosages [64]. A successful cell cycle exit has been associated with androgens' enhanced p27 levels [65]. Studies have revealed that the level of p27 in prostate cancer cells is mainly regulated by the ubiquitin-proteasome system by means of the ubiquitin ligase SCF (SKP1/F-box) SKP2 complex [66, 67]. Androgens specifically modulate the degradation of p27 by SKP2, which in turn raises and stabilizes its levels [68]. Androgens inhibit SKP2 levels in an AR-dependent manner, which stops SKP2 from ubiquitylating and degrading phosphorylated p27 at Thr187 [67, 69, 70]. Nevertheless, SKP2 overexpression is sufficient to overcome androgen-induced G1 block but can override androgen-induced p27 accumulation and facilitate entrance into the S phase [71].
Another plausible mediator of the androgen-regulated p27 is c-myc [72]. Research has shown that c-myc overexpression increased p27 proteolysis and reduced p27's cell cycle-inhibiting effect [73- 75]. Androgens at levels that inhibit cell development in some AR-dependent cell lines decrease c-myc activity, suggesting that the androgenic control of c-myc levels may account for a significant portion of p27 up-regulation [39, 76].
P21
p21 (WAF1/CIP1) is a member of the CDK inhibitors and has been identified as an AR target gene [77-79]. Several studies have demonstrated that p21 induces an apoptotic effect in prostate cancer [39]. This was noted in PC-3 (AR2) cells [30], 104-R2 cells [80], and AR-positive LNCaP-fast growth colony (FGC) cells [81], but not in AR-negative PC-3 cells [82], indicating AR involvement. By selectively binding AR to the consensus ARE, which is situated at the -200 bp in the proximal segment of the p21 promoter, androgens have been demonstrated to raise the levels of p21 transcription [82]. Furthermore, research has demonstrated that the androgenic increase of p21 expression is mediated by the formation of a complex between AR and Specificity Protein 1 (SP1), particularly the SP1-3, which is situated in the p21 promoter [83]. It is hypothesized that prostate cancer cells' apoptotic response will be strengthened by this p21 overexpression, which is dependent on A/AR.
Breast cancer susceptibility gene 1
In addition to its role as a tumor suppressor [84, 85], breast cancer susceptibility gene 1 (BRCA 1) 1 is a key regulator in androgen-induced apoptosis. Previous studies have shown that BRCA 1 stimulates AR transactivation activity by binding directly with AR through two intact pockets within BRCA 1. It is revealed that this binding is ligand-dependent. It has been noted that while DHT or BRCA 1 by themselves can lower the number of cells, their simultaneous addition can raise the proportion of dead cells [84–87]. This impact was seen in DU145, PC-3, and LNCaP cells that had been stably transfected with AR and BRCA 1 [84, 88]. By serving as a link between androgens/AR and RNA polymerase I (transcriptional machinery), BRCA 1 is crucial in mediating the activation of AR target genes, such as p21, that are involved in cell cycle regulation [84, 89].
Protein kinase C
Different cellular responses are known to be regulated by pro-apoptotic Protein Kinase C (PKC) isoenzymes. Protein kinase C delta (PKCδ) is one member of the PKC family that has been demonstrated to cause apoptosis in cells [90-92]. In the presence of androgen, it is hypothesized that AR binds to the ARE, which is situated at 4.7 kb of the human PKCδ gene promoter, to upregulate PKCδ mRNA and protein levels in prostate cancer cells [93]. Significantly, lower levels of PKCδ are observed in LNCaP under androgen deprivation or castration, which is accompanied by the development of resistance to therapies that induce apoptosis [94]. To make androgen-dependent prostate cancer cells more susceptible to PKC's activation of apoptosis, an androgenic pretreatment is therefore necessary. Thus, it is possible to speculate that one of the main factors influencing the apoptotic response in prostate cancer is the up-regulation and modulation of PKCδ by androgens in an AR-dependent way.
TRAIL and TNF-α receptors
TRAIL and TNF-α, popularly referred to as death receptors, are members of the TNF-R family. When activated by a ligand, these receptors cause either cell division, proliferation, or apoptosis [60, 95]. TNF-α threshold levels are a decisive factor in cell cycle control [60]. TNF-α induces apoptosis at greater levels, whereas at lower levels it activates the NF-kB pro-survival pathway to inhibit apoptosis [96]. Androgens, in addition to the androgen-dependent cell lines LNCaP and CWR22RV1, made them more susceptible to TNF-α-induced apoptosis [97]. However, without androgens, the AR-expressing cells became less sensitive. Conversely, even with the testosterone supplementation to the media, the androgen-independent cell lines PC-3, DU145, and JCA-1 demonstrated insensitivity to TNF-α-induced apoptosis [98, 99]. One theory was that this resistance resulted from low levels of TNF stimulating anti-apoptotic genes triggered by NF-kB [98]. It was demonstrated that AR might lessen this resistance in a ligand-dependent fashion by upregulating TNF-α expression and inhibiting NF-kB activity, which would increase the sensitivity of cells to TNF-α-mediated apoptosis [100]. Thus, androgens can affect TNF-α mediated apoptosis.
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a member of the TNF superfamily that may inhibit cancer [101-103]. It has been demonstrated that agonistic antibodies targeting TRAIL receptors exhibit strong anti-tumor action by inducing apoptosis in mouse xenograft models [104]. The inhibition of the Akt/PI3k pathway and modification of caspase 8 activity downstream of the death-inducing signaling complex (DISC) underlie TRAIL-dependent stimulation of apoptosis [101, 105]. Research has shown that androgen ablation in the presence of a PI3k/Akt inhibitor made them resistant to TRAIL treatment [106]. This resistance was linked to lower levels of TRAIL-R1 and TRAIL-R2 in PCa as well as the inability to produce TRAIL-DISC in the absence of androgens [107]. However, in the presence of androgens, the cells' sensitivity to TRAIL-induced apoptosis was fully restored, indicating that androgens enhance TRAIL's ability to cause DISC formation [108]. This, in turn, increases the expression of TRAIL R1 and TRAIL R2, making the cells more susceptible to apoptosis [109]. Consequently, it is possible that sensitivity to TRAIL may improve treatment and, in turn, the clinical management of prostate cancer by combining anti-cancer therapy with androgens.
Epidermal growth factor
According to reports, individuals with elevated levels of the epidermal growth factor receptor (EGFR) exhibit a decreased AR staining index and a heightened likelihood of recurrence [110, 111]. Research has shown that AR modifies EGFR's phosphorylated state and signaling in response to EGF [112-114]. By decreasing the tyrosine phosphorylation of the β4 subunit of integrin α6β4 in PC-3 AR cells, AR reduced the co-localization between EGFR and integrin α6β4v in a ligand-dependent manner. This resulted in a less malignant and more differentiated phenotype in androgen-sensitive prostate cancer cells as well as those transfected with AR [110]. According to recent research, AR inhibits EGFR tyrosine kinase activity by interacting with plasma membrane-associated proteins involved in EGFR signaling, including caveolin-1 and the proto-oncogene tyrosine-protein kinase (Src kinase) family [115]. Furthermore, it has been demonstrated that androgens lessen PC-3AR cells' internalization of EGFR, which lowers the amount of active EGFR inside the cells [113, 115, 116]. All of these results suggested that the deregulation of growth factor signaling pathways in prostate cancer may lead to new and promising therapeutic targets.
C-myc
Numerous studies have reported that an increase in c-myc expression during the early stages of prostate cancer stimulates antiapoptotic genes that contribute to the disease's aggressiveness [117, 118]. It has been shown that the AR, reduces c-myc expression levels in 104-R1 and LNCaP cells in a ligand-dependent manner, resulting in a more differentiated phenotype with an increase in apoptotic indicators [119, 120]. Research findings indicate that castrated rats treated with anti-androgen had higher levels of c-myc mRNA than uncastrated rats or those administered with androgen [121]. Further research is required to determine whether AR binding to its putative binding site in the c-myc gene's first intron controls the expression of the gene and whether this binding can change the rate at which AR regulates the c-myc transcriptional pathway [119].
Nuclear factor kappa B
It has been reported that nuclear factor kappa B (NF-kB) induces the expression of target genes that contribute to tumor progression. These genes include immuno-regulatory, inflammatory, and antiapoptotic genes as well as genes that regulate cell proliferation [122]. Adverse crosstalk between the AR and NF-kB pathways has been suggested by the continuous activation of NF-kB reported in the androgen-resistant DU-145, PC-3, and Du-Pro cell lines, and the extremely low activity of NF-kB observed in the androgen-responsive LNCaP cells [123-127]. It has been proposed that NF-kB excessive expressions shield LNCaP-RelAp65 (LNCaP cells overexpressing NF-kB) cells from apoptosis caused by androgen treatment [128, 129]. IkBα is the inhibitory protein that keeps NF-kB dormant in the cytoplasm. NF-kB can be translocated into the nucleus through the phosphorylation of IkBα by the IκB kinase complex (IKK) in response to various stimuli [130]. Research has shown that in prostate cancer, comparatively high levels of NF-kB located in the nucleus are linked to a poor prognosis and recurring tumors [131, 132]. It has been demonstrated that AR, through a ligand-dependent manner, down-regulates IκB kinase (IKK) expression and reduces the phosphorylation of the IkBα protein [129]. This in turn reduces NF-kB's constitutive activity in prostate cancer and its transfer to the nucleus. Thus, resuming androgen levels during intermittent androgen therapy may therefore make tumor cells more sensitive to proapoptotic substances, which could increase the efficacy of the treatment.
B cell lymphoma 2
The potent pro-survival protein B cell lymphoma 2 (Bcl-2), which is the mammalian counterpart of ced-9, is frequently overexpressed in aggressive and metastatic prostate cancer cells [133, 134]. When androgen ablation occurs, as in the case of androgen-independent (AI) prostate cancer, elevated levels of Bcl-2 have been recorded [135]. This could be partially caused by androgens' inhibitory loss of Bcl-2 [136, 137]. The castrated rats' advancement toward the refractory state was postponed and their Bcl-2 levels were effectively lowered by the administration of androgens. Increased expression of death promoters may be involved in androgen-mediated modulation of Bcl-2 levels [138]. The increasing amounts of Bax caused by androgens create heterodimers with Bcl-2 neutralizing its antiapoptotic effects [139-142]. Bcl2 must be in an active, dephosphorylated state in order to form homodimers with other death inhibitors [143]. Phosphorylation of Bcl-2 by JNK (Jun N-terminal kinase) activation may aid in its deactivation [144, 145]. Therefore, it would be interesting to investigate how A/AR contributes to the inactivation of Bcl-2 in prostate cancer via upregulating JNK.
E2F transcription factor 1
E2F transcription factor 1 (E2F1) plays a role in the androgenic reduction of Bcl-2 expression. Increased Bcl-2 protein and mRNA levels in LNCaP cells are a result of ectopic expression of E2F1. Research has indicated that there is a reciprocal relationship between the AR and E2F pathway [146, 147]. E2F1 protein levels have been found to be downregulated by androgens/AR, as is the degree to which E2F1 binds to the Bcl-2 promoter. It has been shown that LNCaP and PC-3AR cells express fewer Bcl-2 and other E2F1-regulated genes [146, 147]. Thus, it stands to reason that changed E2F1 levels could account for the treatment resistance linked to AR dysregulation.
THERAPEUTIC ADVANCES AND PROSPECTS OF ANDROGEN/AR
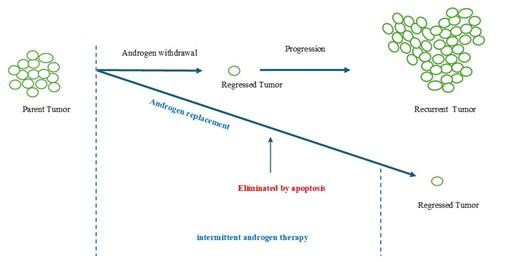
For advanced prostate cancer, castration is an effective therapy option, either by itself or in conjunction with androgen ablation (Figure 3). After responding favorably to treatment at first, patients progress to an androgen-refractory tumor, a hormone-resistant stage that is very likely to propagate. Additionally, androgen insensitivity has been linked to modifications in the regulation of apoptotic pathways. One important sign of incurable oncogenic transformation seems to be a decrease in apoptosis sensitivity or an increase in resistance. It has been suggested that restoring testosterone levels during intermittent androgen therapy can alter the way tumor cells proliferate and increase their susceptibility to pro-apoptotic agents.
The role of androgen/AR in apoptosis in advanced prostate cancer is revealed by intermittent androgen deprivation therapy (IAD). Compared to continuous androgen deprivation therapy (ADT), IAD aids patients with their symptoms and slows the growth of tumors [148]. Moreover, IAD therapy can lessen the development of castration-resistant prostate cancer in men with prostate cancer who have undergone primary treatment, merely biochemically progressed, and were chosen for ADT [149]. A/AR's apoptotic function during IAD therapy can lessen the adverse effects of ADT, which worsen as therapy duration increases. In addition, IAD has several other benefits such as postponing androgen independence, lowering therapy morbidity, improving life satisfaction (e.g., sexual desire, urinary discomfort), maintaining bone density, and is cost-effective [150]. The existing clinical research shows that both treatment approaches were safe and effective, although IAD did not surpass continuous ADT in minimizing prostate cancer's progression and mortality. Intermittent androgen deprivation therapy was not inferior to continuous androgen deprivation therapy regarding overall survival. Intermittent therapy appeared to be associated with better quality-of-life outcomes. The utilization of intermittent androgen deprivation may be recommended for individuals with recurrent or metastatic prostate cancer [151].
CONCLUSION
Therapeutic approaches to selectively induce apoptosis by manipulating apoptotic pathways are being explored in a variety of clinical circumstances, and such approaches have been suggested as possible strategies to enhance intermittent androgen therapy in the treatment of prostate cancer. In this review, we slight the mechanism of steroid hormones such androgen/AR pathway in the mechanism inducing apoptosis in prostate cancer.
ACKNOWLEDGEMENTS
None.
AUTHOR CONTRIBUTIONS
This work involved collaboration among all the authors. SA and SMA designed the outline, wrote the initial draft, and reviewed the scientific contents described in the manuscript. All the authors have approved the final version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Lang SH, Frame FM, et al. Prostate cancer stem cells. The Journal of pathology. 2009; 217(2):299-306.
- [2]Kirby RS. Prostate cancer. 7th ed. ed. Abingdon: Abingdon: Health Press. 2012
- [3]Swami U, McFarland TR, Nussenzveig R, Agarwal N. Advanced Prostate Cancer: Treatment Advances and Future Directions. Trends Cancer. 2020;6(8):702-715
- [4]Lim J, Onozawa M, et al. Recent trend of androgen deprivation therapy in newly diagnosed prostate cancer patients: Comparing between high‐and middle‐income Asian countries. Cancer Sci. 2021;112(6):2071-80.
- [5]Merkens L, Sailer V, et al. Aggressive variants of prostate cancer: underlying mechanisms of neuroendocrine transdifferentiation. J Exp Clin Cancer Res. 2022;41(1):46.
- [6]Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005;23(32):8253-61.
- [7]Takayama KI, Inoue S. Transcriptional network of androgen receptor in prostate cancer progression. Int J Urol .2013;20(8):756-68.
- [8]Davey RA, Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. 2016;37(1):3.
- [9]Messner EA, Steele TM, et al. The androgen receptor in prostate cancer: Effect of structure, ligands and spliced variants on therapy. Biomedicines. 2020;8(10):422.
- [10]Antonarakis ES, Chandhasin C, et al. Targeting the N-terminal domain of the androgen receptor: a new approach for the treatment of advanced prostate cancer. The oncologist. 2016; 21(12):1427-35.
- [11]Helsen C, Dubois V, et al. Evidence for DNA-binding domain–ligand-binding domain communications in the androgen receptor. Mol Cell Biol. 2012;32(15):3033–3043.
- [12]Ratajczak W, Lubkowski M, et al. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. Int J Mol Sci. 2022;23(2):897.
- [13]Yu X, Yi P, et al. Structural insights of transcriptionally active, full-length androgen receptor coactivator complexes. Mol. Cell. 2020;79(5):812-23.
- [14]Ozyerli-Goknar E, Bagci-Onder T. Epigenetic deregulation of apoptosis in cancers. Cancers. 2021;13(13):3210.
- [15]Singh R, Letai A, et al. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019;20(3):175-93.
- [16]Neophytou CM, Trougakos IP, et al. Apoptosis deregulation and the development of cancer multi-drug resistance. Cancers. 2021;13(17):4363.
- [17]Abd. Wahab NA, H. Lajis N, et al. Mechanism of anti-cancer activity of curcumin on androgen-dependent and androgen-independent prostate cancer. Nutrients. 2020;12(3):679.
- [18]Maitland NJ. Resistance to antiandrogens in prostate cancer: is it inevitable, intrinsic or induced? Cancers. 2021;13(2):327.
- [19]Cohen MB, Rokhlin OW. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J Cell Biochem. 2009;106(3):363-71.
- [20]Klotz LH, Herr HW, et al. Intermittent endocrine therapy for advanced prostate cancer. Cancer. 1986;58:2546-50.
- [21]Karantanos T, Corn PG, et al. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013; 32(49):5501-11.
- [22]Joly-Pharaboz MO, Ruffion A, et al. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J Steroid Biochem Mol Biol. 2000;73(5):237-49.
- [23]Chuu CP, Hiipakka RA, et al. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res. 2005;65(6):2082-4.
- [24]Bruckheimer EM, Kyprianou N. Dihydrotestosterone enhances transforming growth factor-β-induced apoptosis in hormone-sensitive prostate cancer cells. Endocrinol. 2001;142(6):2419-26.
- [25]Kurek R, Renneberg H, et al. Intermittent complete androgen blockade in PSA relapse after radical prostatectomy and incidental prostate cancer. Eur. Urol. 1998;35(Suppl. 1):27-31.
- [26]Tunn UW, Bruchovsky N, et al. Intermittierende Androgendeprivation. Der Urologe A. 2000; 39:9-13.
- [27]Vachalovský V, Dvoracek J. Intermittent androgen blockade in prostatic carcinoma. Casopis Lekaru Ceskych. 2002;141(21):669-672.
- [28]Kaighn ME, Narayan KS, et al. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest. Urol.1979;17(1):16-23.
- [29]Bonaccorsi L, Carloni V, et al. Androgen receptor expression in prostate carcinoma cells suppresses α6β4 integrin-mediated invasive phenotype. Endocrinol. 2000;141(9):3172-82.
- [30]Heisler LE, Evangelou A, et al. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol. Cell. Endocrinol. 1997;126(1):59-73.
- [31]Yuan S, Trachtenberg J, et al. Androgen-induced inhibition of cell proliferation in an androgen-insensitive prostate cancer cell line (PC-3) transfected with a human androgen receptor complementary DNA. Cancer Res. 1993;53(6):1304-11.
- [32]Mickey DD, Stone KR, et al. Characterization of a human prostate adenocarcinoma cell line (DU 145) as a monolayer culture and as a solid tumor in athymic mice. Prog. Clin. Biol. Res. 1980; 37:67-84.
- [33]Stone KR, Mickey DD, et al. Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer. 1978;21(3):274-81.
- [34]Yuan S, Trachtenberg J, et al. Androgen-induced inhibition of cell proliferation in an androgen-insensitive prostate cancer cell line (PC-3) transfected with a human androgen receptor complementary DNA. Cancer Res. 1993;53(6):1304-11.
- [35]Hasenson M, Pousette Å, et al. Effect of hormones on growth and ATP content of a human prostatic carcinoma cell line, LNCaP‐r. The Prostate. 1985;7(2):183-94.
- [36]Kokontis J, Takakura K, et al. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 1994; 54(6):1566-73.
- [37]Choo CK, Ling MT, et al. Immortalization of human prostate epithelial cells by HPV 16 E6/E7 open reading frames. The Prostate. 1999;40(3):150-8.
- [38]Altuwaijri S, Wu CC, et al. Expression of human AR cDNA driven by its own promoter results in mild promotion, but not suppression, of growth in human prostate cancer PC‐3 cells. Asian J. Androl. 2007;9(2):181-8.
- [39]Kokontis JM, Hay N, et al. Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol Endocrinol. 1998;12(7):941-53.
- [40]Wang X, Deng H, et al. Induction of androgen receptor-dependent apoptosis in prostate cancer cells by the retinoblastoma protein. Cancer Res. 2004;15;64(4):1377-85.
- [41]Altuwaijri S, Lin HK, et al. Interruption of nuclear factor κB signaling by the androgen receptor facilitates 12-O-tetradecanoylphorbolacetate-induced apoptosis in androgen-sensitive prostate cancer LNCaP cells. Cancer Res. 2003;63(21):7106-12.
- [42]Ling MT, Chan KW, et al. Androgen induces differentiation of a human papillomavirus 16 E6/E7 immortalized prostate epithelial cell line. J. Endocrinol. 2001;170(1):287-96.
- [43]Vélez-Cruz R, Johnson DG. The retinoblastoma (RB) tumor suppressor: pushing back against genome instability on multiple fronts. Int. J. Mol. Sci. 2017;18(8):1776.
- [44]Day ML, Foster RG, et al. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J. Biol. Chem. 1997;272(13):8125-8.
- [45]Sharma A, Yeow WS, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120(12):4478-92.
- [46]Yeh S, Miyamoto H, et al. Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochem Biophys Res. 1998; 248(2):361-7.
- [47]Chen PL, Scully P, et al. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell. 1989;58(6):1193-8.
- [48]Huang H, Zegarra-Moro OL, et al. Androgens repress Bcl-2 expression via activation of the retinoblastoma (RB) protein in prostate cancer cells. Oncogene. 2004;23(12):2161-76.
- [49]Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat. Rev. Cancer. 2008;8(9):714-24.
- [50]Qian S, Wei Z, et al. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022;12:985363.
- [51]Jeong SH, Kim HH, et al. The Apoptosis in Prostate Cancer of Three Flavones Selected as Therapeutic Candidate Models. Int. J. Mol. Sci. 2023;24(11):9240.
- [52]Gregory GL, Copple IM. Modulating the expression of tumor suppressor genes using activating oligonucleotide technologies as a therapeutic approach in cancer. Mol Ther Nucleic Acids. 2023;31:211-23.
- [53]Gross A, Jockel J, et al. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17(14):3878-85.
- [54]Lin Y, Kokontis J, et al. Androgen and its receptor promote Bax-mediated apoptosis. Mol Cell Biol. 2006;26(5),1908–1916.
- [55]Russo A, Cardile V, et al. Involvement of Bax and Bcl-2 in induction of apoptosis by essential oils of three Lebanese Salvia species in human prostate cancer cells. Int. J. Mol. Sci. 2018; 19(1):292.
- [56]Andriani F, Nan B, et al. Use of the probasin promoter ARR2PB to express Bax in androgen receptor-positive prostate cancer cells. J Natl Cancer Inst. 2001;93(17):1314-24.
- [57]Lowe SL, Rubinchik S, et al. Prostate-specific expression of Bax delivered by an adenoviral vector induces apoptosis in LNCaP prostate cancer cells. Gene Ther. 2001;8(18):1363-71.
- [58]Dostert C, Grusdat M, et al. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol Rev. 2019;99(1):115-160.
- [59]Lorenzo PI, Arnoldussen YJ, et al. Molecular mechanisms of apoptosis in prostate cancer. Crit Rev Oncog. 2007;13(1):1-38.
- [60]Lee J, Kim SS. The function of p27KIP1 during tumor development. Exp Mol Med. 2009; 41(11):765-71.
- [61]Chuu CP, Kokontis JM, et al. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci. 2011;18:1-2.
- [62]Blain SW, Scher HI, et al. p27 as a target for cancer therapeutics. Cancer cell. 2003;3(2):111-5.
- [63]Fang Z, Zhang T, et al. Androgen receptor enhances p27 degradation in prostate cancer cells through rapid and selective TORC2 activation. J. Biol. Chem. 2012;287(3):2090-8.
- [64]Ben-Salem S, Venkadakrishnan VB, et al. Novel insights in cell cycle dysregulation during prostate cancer progression. Endocrine-related cancer. 2021;28(6):R141-55.
- [65]Rico-Bautista E, Yang CC, et al. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biol 2010;8:1-8.
- [66]Spruck C, Strohmaier H, Watson M, et al. A CDK-independent function of mammalian Cks1: targeting of SCFSkp2 to the CDK inhibitor p27Kip1. Molecular cell. 2001;7(3):639-50.
- [67]Michmerhuizen AR, Spratt DE, et al. ARe we there yet? Understanding androgen receptor signaling in breast cancer. NPJ breast cancer. 2020;6(1):47.
- [68]Song K, Wang H, et al. Androgenic control of transforming growth factor-β signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-β receptor II. Cancer Res. 2008;68(19):8173-82.
- [69]Montagnoli A, Fiore F, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes & development 1999;13(9):1181-9.
- [70]Lu L, Schulz H, et al. The F-box protein SKP2 mediates androgen control of p27 stability in LNCaP human prostate cancer cells. BMC cell Biol. 2002;3:1-3.
- [71]Shtivelman E, Beer TM, et al. Molecular pathways and targets in prostate cancer. Oncotarget. 2014;5(17):7217.
- [72]Vlach J, Hennecke S, et al. Growth arrest by the cyclin‐dependent kinase inhibitor p27Kip1 is abrogated by c‐Myc. The EMBO J. 1996;15(23):6595-604.
- [73]Rohan JN, Weigel NL. 1α, 25-dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4-2 prostate cancer cells. Endocrinol. 2009; 150(5):2046-54.
- [74]Steiner P, Philipp A, Lukas J, et al. Identification of a Myc‐dependent step during the formation of active G1 cyclin‐cdk complexes. The EMBO J. 1995;14(19):4814-26.
- [75]Chatterjee P, Schweizer MT, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor–mediated DNA damage J Clin Invest. 2019;129(10):4245-60.
- [76]Lu S, Liu M, et al. Androgen regulation of the cyclin-dependent kinase inhibitor p21 gene through an androgen response element in the proximal promoter. Mol Endocrinol. 1999; 13(3):376-84.
- [77]Harper JW, Adami GR, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805-16.
- [78]Xiong Y, Hannon GJ, et al. p21 is a universal inhibitor of cyclin kinases. Nature 1993; 366:701–704
- [79]Shamloo B, Usluer S. p21 in cancer research. Cancers. 2019;11(8):1178.
- [80]Lu S, Tsai SY, et al. Regulation of androgen-dependent prostatic cancer cell growth: androgen regulation of CDK2, CDK4, and CKI p16 genes. Cancer Res. 1997;57(20):4511-6.
- [81]Kokontis JM, Lin HP, et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS One. 2014;9(10):e109170.
- [82]Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9(6):400-14.
- [83]Yeh S, Hu YC, et al. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc. Natl. Acad. Sci. 2000;97(21):11256-61.
- [84]Harkin DP, Bean JM, et al. Induction of GADD45 and JNK/SAPK dependent apoptosis following inducible expression of BRCA1.Cell. 1999;97(5):575-86.
- [85]MacLachlan TK, Somasundaram K, et al. BRCA1 effects on the cell cycle and the DNA damage response are linked to altered gene expression. J Biol Chem. 2000;275(4):2777-85.
- [86]Somasundaram K, Zhang HB, et al. Arrest of the cell cycle by the tumor-suppressor BRCA1 requires the CDK-inhibitor p21/WAF1/CIP1. Nature. 1997;389:187–190.
- [87]Cleutjens KB, van der Korput HA, et al. An Androgen Response Element in a Far Upstream Enhancer Region Is Essential for High, Androgen-Regulated Activity of the Prostate-Specific Antigen Promoter. Mol Endocrinol. 1997;11:148–161.
- [88]Anderson SF, Schlegel BP, et al. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet 1998;19(3):254–256
- [89]Kawano T, Inokuchi J, et al. Protein kinase C (PKC) isozymes as diagnostic and prognostic biomarkers and therapeutic targets for cancer. Cancers. 2022;14(21):5425.
- [90]Kang JH. Protein kinase C (PKC) isozymes and cancer. New J. Sci. 2014; (1);231418.
- [91]Black JD, Affandi T, et al. PKCα and PKCδ: friends and rivals. J. Biol. Chem. 2022;298(8).
- [92]Gavrielides MV, Gonzalez-Guerrico AM, et al. Androgens regulate protein kinase Cδ transcription and modulate its apoptotic function in prostate cancer cells. Cancer Res. 2006; 66(24):11792-801.
- [93]Montalvo L, Sanchez-Chapado M, et al. Regulation of the expression of protein kinase C isoenzymes in rat ventral prostate: effects of age, castration and flutamide treatment. Life Sci. 2002;71:2257–66.
- [94]Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811.
- [95]Koliaki C, Katsilambros N. Repositioning the role of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) on the TRAIL to the development of diabetes mellitus: An update of experimental and clinical evidence. Int. J. Mol. Sci. 2022;23(6):3225.
- [96]Ting AT, Bertrand MJ. More to life than NF-κB in TNFR1 signaling. Trends Immunol. 2016; 37(8):535-45.
- [97]Shankar E, Song K, et al. A signaling network controlling androgenic repression of c-Fos protein in prostate adenocarcinoma cells. J. Biol. Chem. 2016; 291(11):5512-26.
- [98]Rokhlin OW, Taghiyev AF, et al. Androgen regulates apoptosis induced by TNFR family ligands via multiple signaling pathways in LNCaP. Oncogene. 2005;24(45):6773-84.
- [99]Yu H, Lin L, et al. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct and Tar Ther. 2020;5(1):209.
- [100]Bucur O, Ray S, et al. APO2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in prostate cancer therapy. Front Biosci. 2006;11:1549-68.
- [101]Bouralexis S, Findlay DM, et al. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 2005;10:35–51.
- [102]Diaz Arguello OA, Haisma HJ. Apoptosis-inducing TNF superfamily ligands for cancer therapy. Cancers. 2021;13(7):1543.
- [103]Fulda S, Debatin KM. Signaling through death receptors in cancer therapy. Curr Opin Pharmacol. 2004;4(4):327-32.
- [104]Tummers B, Green DR. Caspase‐8: regulating life and death. Immunol. Rev. 2017; 277(1):76-89.
- [105]Ehsani M, David FO, et al. Androgen receptor-dependent mechanisms mediating drug resistance in prostate cancer. Cancers. 2021;13(7):1534.
- [106]Staniek J, Lorenzetti R, et al. TRAIL-R1 and TRAIL-R2 mediate TRAIL-dependent apoptosis in activated primary human B lymphocytes. Front Immunol. 2019;10:951.
- [107]Farooqi AA, Gadaleta CD, et al. Restoring trail induced apoptosis using naturopathy. Hercules joins hand with nature to triumph over lernaean hydra. Curr Genomics. 2017;18(1):27.
- [108]Sarhan D, D’Arcy P, et al. Regulation of TRAIL-receptor expression by the ubiquitin-proteasome system. Int J Mol Sci. 2014;15(10):18557-73.
- [109]Schäfer W, Funke PJ, et al. Intensity of androgen and epidermal growth factor receptor immunoreactivity in samples of radical prostatectomy as prognostic indicator: correlation with clinical data of long-term observations. Urol. J. 2006;176(2):532-7.
- [110]Zhimin Ji, Lili Yang, et al. Correlation of epidermal growth factor receptor (EGFR), androgen receptor (AR) and 14-3-3 sigma expression in breast cancer. Int. J Clin Exp. Pathol. 2017; 10(10):10419-30.
- [111]Chuu CP, Kokontis JM, et al. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci. 2011;18:1-2.
- [112]Bonaccorsi L, Muratori M, et al. The androgen receptor associates with the epidermal growth factor receptor in androgen-sensitive prostate cancer cells. Steroids. 2004;69(8-9):549-52.
- [113]Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9(5):52.
- [114]Bonaccorsi L, Carloni V, et al. EGF receptor (EGFR) signaling promoting invasion is disrupted in androgen‐sensitive prostate cancer cells by an interaction between EGFR and androgen receptor (AR). Int. J. Cancer. 2004;112(1):78-86.
- [115]Bonaccorsi L, Nosi D, et al. Altered endocytosis of epidermal growth factor receptor in androgen receptor positive prostate cancer cell lines. J. Mol. Endocrinol 2007;38(1):51-66.
- [116]Koh CM, Bieberich CJ, et al. MYC and Prostate Cancer. Genes Cancer. 2010;(6):617-28.
- [117]Yang G, Timme TL, et al. Combined c‐Myc and caveolin‐1 expression in human prostate carcinoma predicts prostate carcinoma progression. Cancer: Interdiscip Int J Am Cancer Soc. 2005;103(6):1186-94.
- [118]Williams K, Fernandez S, et al. Unopposed c‐MYC expression in benign prostatic epithelium causes a cancer phenotype. The Prostate. 2005;63(4):369-84.
- [119]Ellwood-Yen K, Graeber TG, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer cell. 2003;4(3):223-38.
- [120]Wolf DA, Kohlhuber F, et al. Transcriptional down-regulation of c-myc in human prostate carcinoma cells by the synthetic androgen mibolerone. Br. J. Cancer. 1992;65(3):376-82.
- [121]Chuu CP, Kokontis JM, et al. Androgen suppresses proliferation of castration‐resistant LNCaP 104‐R2 prostate cancer cells through androgen receptor, Skp2, and c‐Myc. Cancer Sci. 2011;102(11):2022-8.
- [122]Bai S, Cao S, et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene. 2019;38(25):4977-89.
- [123]Karin M, Cao Y, et al. NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer. 2002;2(4):301-10.
- [124]Suh J, Rabson AB. NF-kB activation in human prostate cancer: important mediator or epiphenomenon? J Cell Biochem. 2004;91:100 –17.
- [125]Chen CD, Sawyers CL. NF-κB activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol Cell Biol. 2002;22(8):2862-70.
- [126]Alimirah F, Chen J, et al. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS letters. 2006;580(9):2294-300.
- [127]Péant B, Diallo JS, et al. Regulation of IκB kinase ε expression by the androgen receptor and the nuclear factor-κB transcription factor in prostate cancer. Mol Cancer Res. 2007;5(1):87-94.
- [128]Khurana N, Sikka SC. Targeting crosstalk between Nrf-2, NF-κB and androgen receptor signaling in prostate cancer. Cancers. 2018;10(10):352.
- [129]Basílio J, Hochreiter B, et al. Antagonistic functions of androgen receptor and NF-κB in prostate cancer—Experimental and computational analyses. Cancers. 2022;14(24):6164.
- [130]Palvimo JJ, Reinikainen P, et al. Mutual transcriptional interference between RelA and androgen receptor. J. Biol. Chem. 1996;271(39):24151-6.
- [131]Hayden MS, Ghosh S. Signaling to NF-kB. Genes Dev. 2004;18:219–224.
- [132]Ismail A H, Lessard L, et al. Expression of NF‐κB in prostate cancer lymph node metastases. The Prostate. 2004;58(3):308-13.
- [133]Zhang T, Ma C, et al. NF‐κB signaling in inflammation and cancer. MedComm. 2021; 2(4):618-53.
- [134]Waltregny D, Leav I, et al. Androgen-driven prostate epithelial cell proliferation and differentiation in vivo involve the regulation of p27. Mol Endocrinol. 2001;15(5):765-82.
- [135]Kirkin V, Joos S, et al. The role of Bcl-2 family members in tumorigenesis. Biochim. Biophys. Acta, Mol. Cell Res. 2004;1644(2-3):229-49.
- [136]Lin Y, Fukuchi J, et al. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007;17(6):531-6.
- [137]Cardillo M, Berchem G, et al. Resistance to apoptosis and up regulation of Bcl-2 in benign prostatic hyperplasia after androgen deprivation. J Urol. 1997;158(1):212-6.
- [138]Xiang Z, Sun Y, et al. Suppressing BCL-XL increased the high dose androgens therapeutic effect to better induce the Enzalutamide-resistant prostate cancer autophagic cell death. Cell Death Dis. 2021;12(1):68.
- [139]Lapointe J, Fournier A, et al. Androgens down-regulate bcl-2 protooncogene expression in ZR-75-1 human breast cancer cells. Endocrinol. 1999;140(1):416-21.
- [140]Oltval ZN, Milliman CL, et al. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell. 1993;74(4):609-19.
- [141]Antonsson B, Conti F, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277(5324):370-2.
- [142]Abd. Wahab NA, H. Lajis N, et al. Mechanism of anti-cancer activity of curcumin on androgen-dependent and androgen-independent prostate cancer. Nutrients. 2020;12(3):679.
- [143]Wen S, Niu Y, et al. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat. Rev. 2014;40(1):31-40.
- [144]Kale J, Osterlund EJ, et al. BCL-2 family proteins: changing partners in the dance towards death. CELL DEATH DIFFER. 2018;25(1):65-80.
- [145]Yamamoto K, Ichijo H, et al. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol Cell Biol. 1999;19(12):8469-78.
- [146]Sharma A, Yeow WS, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. invest. 2010;120(12):4478-92.
- [147]Thomas S, Quinn BA, et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013;17(1):61-75.
- [148]Perera M, Roberts MJ, et al. Intermittent versus continuous androgen deprivation therapy for advanced prostate cancer. Nat Rev Urol. 2020;17(8):469-481
- [149]Salciccia S, Frisenda M, et al. Intermittent Versus Continuous Androgen Deprivation Therapy for Biochemical Progression After Primary Therapy in Hormone-Sensitive Nonmetastatic Prostate Cancer: Comparative Analysis in Terms of CRPC-M0 Progression. Clin. Genitourin. Cancer. 2024;22(2):74-83.
- [150]Dong Z, Wang H, et al. Intermittent hormone therapy versus continuous hormone therapy for locally advanced prostate cancer: a meta-analysis. The Aging Male. 2015;18(4): 233-237.
- [151]Magnan S, Zarychanski R, et al. Intermittent vs continuous androgen deprivation therapy for prostate cancer: a systematic review and meta-analysis. JAMA Oncol. 2015;1(9):1261-1269.