Prediction of immune pressure on HIV-1 regulatory gene tat by human host through bioinformatics tools
Abstract
In the early establishment of HIV-1 infection, Tat protein plays an essential role in controlling other genes of HIV-1 (e.g., vif, vpr, vpu, nef, gag, pol and env), for viral pathogenesis, while maintaining its polymorphic nature. It is well documented that polymorphism of HIV-1 genome are created to escape immune pressures (e.g. CD8+, CD4+, B, NK-cells and others) by human host, during the course of infection. Over the time those mutations are incorporated or left in the HIV genome as an escaped flag or signature amino acid, and this scenario could be predictable using contemporary bioinformatics tools. Our sequence analysis from global database (LANL) revealed that Tat protein under positive immune pressure as dn/ds >1.5, even though differential immune pressure exists among the HIV-1 subtype. Average entropy score is 0.31, implying the less variable nature of this protein while amino acid variations are higher in C-terminal. Remarkably, the region encompassing by positions 38 to 51 amino acids are relatively conserved, over the year 2009 to 2017 across HIV-1 subtype. Indeed, subtype-specific SNP or signature amino acids were observed in various position of Tat, dominantly in C-terminal end. Our epitope density plot analysis, highlighted CTLs/CD8+ cells play a major role on Tat sequence variation around the globe. Taken together, our analysis illustrated the dynamic nature of polymorphism within HIV-1 proteins, which could be predictable to see the immune-mediated selective pressure by human host on viral genome.
INTRODUCTION
One of the notorious characteristics of HIV-1 virus is tremendous sequence variation, creating highly divergent circulating strains around the globe [1]. Designing an ideal/universal vaccine against HIV-1 has been threatening to the scientific community due to this genetic variability and ability to escape a myriad of host immune responses [2]. Remarkably, HIV-1 creates several mutations in its genome to escape immune-mediated selective pressure (e.g. CD8+, CD4+, NK, B-cells, host factor and others) by the human hosts during the course of infection [3-6]. Although, over the time those mutations are incorporated or left in the HIV sequence as a flag or signature amino acid, when HIV transmit to other human hosts while facing different immune profile and this scenario could be predictable using contemporary bioinformatics tools [3]. To escape or adapt immune pressure, HIV-1 acquire mutation (i.e. synonymous and nonsynonymous) and try to escape host responses while maintaining their fitness effects as minimal as possible.
In the early establishment of HIV-1 infection Tat protein plays an important controlling role of other HIV-1 gene expressions (e.g. vif, vpr, vpu, nef, gag, pol and env ) for viral replication and disease pathogenesis [7, 8]. Despite its important role, it is evident that Tat is also variable proteins in HIV proteome [9], indicating Tat is under immune pressure and evolving to adapt in the respective host system to act as a functional protein.
Several studies have shown that sequence polymorphism of HIV-1 Tat exists among the subtype (B, C, E and BF) of HIV-1 [10]. Interestingly subtype C Tat exhibits greater transcriptional activity in the CD4+ T cell line compared with subtypes B and E and this is because of variations in positions 57 (Ser to Arg) and 63 (Glu to Gln), suggesting the Tat sequence variability has a significant advantage on HIV-1 subtype C replication [10, 11]. Therefore, the adaptation of sequence polymorphism among various HIV-1 subtypes in different subcontinents, highlighting host immune profile may be one of the driving forces of these polymorphisms, which, however, is one of the setbacks of designing effective vaccine so far. Thus, a comprehensive analysis of the dynamics of polymorphisms in HIV-1 proteins is a powerful tool to reveal actual interactions between HIV-1 and the host immune system. Moreover, to decipher HIV-1 tat gene sequence evolution and possible candidate target in vaccination strategy, it is inevitable to see how this gene being imposed with selective pressure by the human host, which could be predicted using publicly open-access portal of HIV sequence and immunology database along with the contemporary bioinformatics tools from Los Alamos National Laboratory (LANL) (https://www.hiv.lanl.gov).
MATERIALS AND METHODS
Sequence database
All the deposited tat gene nucleotide (intact ORF) sequence from the year 2009 to 2017 was retrieved from the Los Alamos National Laboratory (LANL) HIV sequence database. The HIV database contain comprehensive data on HIV genome sequences and allow to access large number of bioinformatical tools that can be used to analyze and visualize the dataset. In brief, from this website (https://www.hiv.lanl.gov), anyone can choose from the options button to define:- [i] Alignment type (web, filtered, subtype, compendium, and consensus/ancestral), [ii] Organism (HIV-1/2 or SIV), [iii] Region of the genome (env, nef, vif, vpr, cpu, and others), [iv] Subtype (A-K and recombinants), [v] DNA/Protein, [vi] Year, [vii] Format (FASTA, Clustal, Phylip, MEGA and others ) and get alignment file on their gene of interest.
Sequence alignment
Retrieved sequence dataset was edited in MEGA7 (Molecular Evolutionary Genetics Analysis) software, an alignment tool for sequence manipulation [12]. Sequences were spilt-up (n=150 to 200/subtype, each country not more than 10 sequences) based on subtype variation (A, B, C, D, F, G, H CRFs (AE, BC and AG) and CPX. Gaps were removed from the above selected multiple sequences in notepad. Finally, the non-align multiple sequence datasets were aligned again with Clustal W in MEGA7 [13] with respect to consensus reference subtype sequence (latest year 2004) from LANL (https://www.hiv.lanl.gov/).
Selective pressure and dn/ds ratio
To find the natural selection pressure on tat gene, dn/ds ratio was used using SNAP v2.1.1 [(synonymous (ds) vs non-synonymous (dn)] tools from LANL, which calculates dn and ds substitution rates based on a set of codon-aligned nucleotide sequences [1]. In briefly, multiple aligned nucleotide sequence as FASTA files were uploaded in synonymous non-synonymous analysis site (https://www.hiv.lanl.gov/) to get the dn and ds values for each 101 position. Subsequently the average value of dn was divided with average ds to get dn/ds ratio. If dn/ds ratio ≥ 1, Positive selection and pathogen try to escape from host immune surveillance while dn/ds ratio < 1, Negative/Purifying selection and pathogen try to adapt to host immune system.
Variability of amino acid sequence of Tat protein
A Shannon entropy score for each position in the Tat protein was calculated to see the extent of amino acid sequence variability [9]. In this application, multiple aligned amino acid sequence as FASTA files from Clustal W (translated to amino acid) were uploaded in the Shannon Entropy-One site to get the frequency of each position as column in a sequence alignment independently. As such, this entropy tool assigns a score to each column that reflects the variability in that column. Finally, all the values for each position of Tat protein (ORF, 110 aa) were processed in excel and prism file for further analysis.
Epitope density plots
HIV molecular immunology database is a summary of all HIV-1 epitopes, which have been reported in the literature, including HIV-1 Cytotoxic (CTL/CD8+), T-helper/CD4+ and Antibody/B-cells epitopes sites. Density plot illustrated the number of reported human HIV-1 (https://www.hiv.lanl.gov/) epitopes spanning each amino acid of the targeted protein. In brief, the number of reported epitopes to each amino acid (aa) position of Tat protein (1-101 aa), from this database was recovered that already been positioning with HIV-1 reference strain HXB2. All the numerical values for T cells (CD4+ and CD8+) and B-cells were transferred to excel file followed by GraphPad Prism to create the figure and analysis, if required.
Statistical analysis
All the graphs and figures were generated using multivariate statistical package GraphPad Prism software (Version 6.0, La Jolla, CA, United States) and Excel. A p value <0.05 indicated by asterisk as significant, otherwise p value >0.05, non-significant using student’s t-test (two-tailed).
RESULTS
Host mediated selective pressure on HIV-1 tat gene
The overall immune pressure on HIV-1 tat gene across the subtype is positive, dn/ds ratio ≥ 1. The average dn/ds value is more than 1.5 over the year from 2009 to 2017 but varied among the subtypes (Figure 1A and 1B). This result suggests that, tat gene of circulating HIV-1 strains around the globe undergoing immune pressure by the human host, because the rate of substitutions at non-silent sites (dn, experienced selection) are higher than the rate of substitutions at silent sites (ds, presumed neutral). In particular, our analysis suggests that tat may experience substantially stronger selective forces, hence selection pressure and dn/ds is qualitatively different for samples drawn from a single population compared to sampled from various or mixed population [14]. Interestingly, only subtype G showed lower dn/ds ratio (mean=1.1) over the year, while other subtypes did not. In addition, the prevalent subtypes responsible for global HIV/AIDS are B (North America, Europe, and Australia) and C (South Africa and South East Asia) [15], have shown statistically significant difference of dn/ds ratio (B, mean=1.71; C, mean=1.49) over the year 2009 to 2017 on tat gene (Figure 1C). Taken together, our analysis suggests that the differential adaptation of HIV-1 tat gene across the globe, due to the variation of human immune profile and imposed pressure.
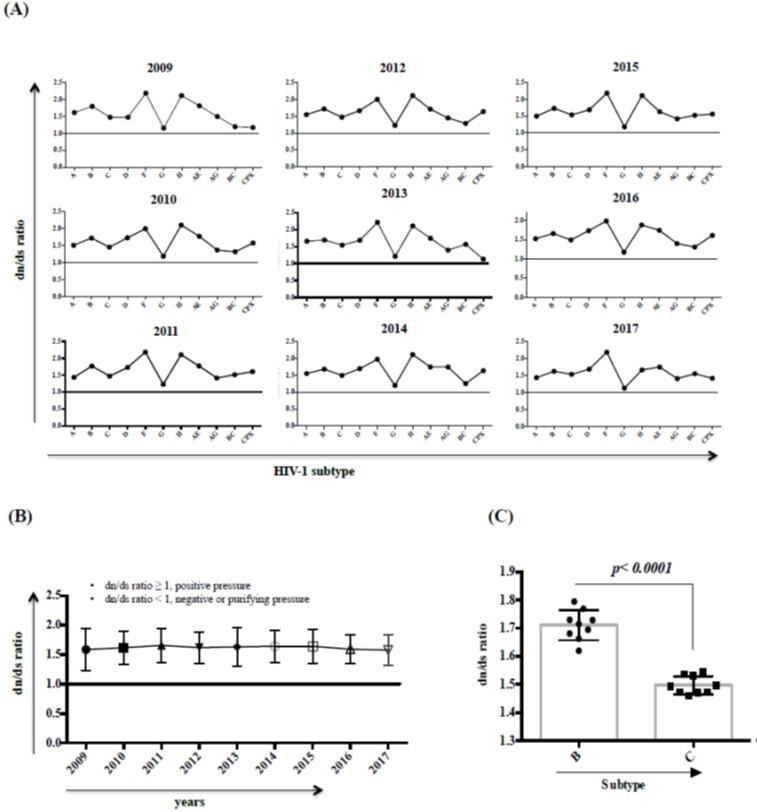
Variability of Tat protein and signature amino acid
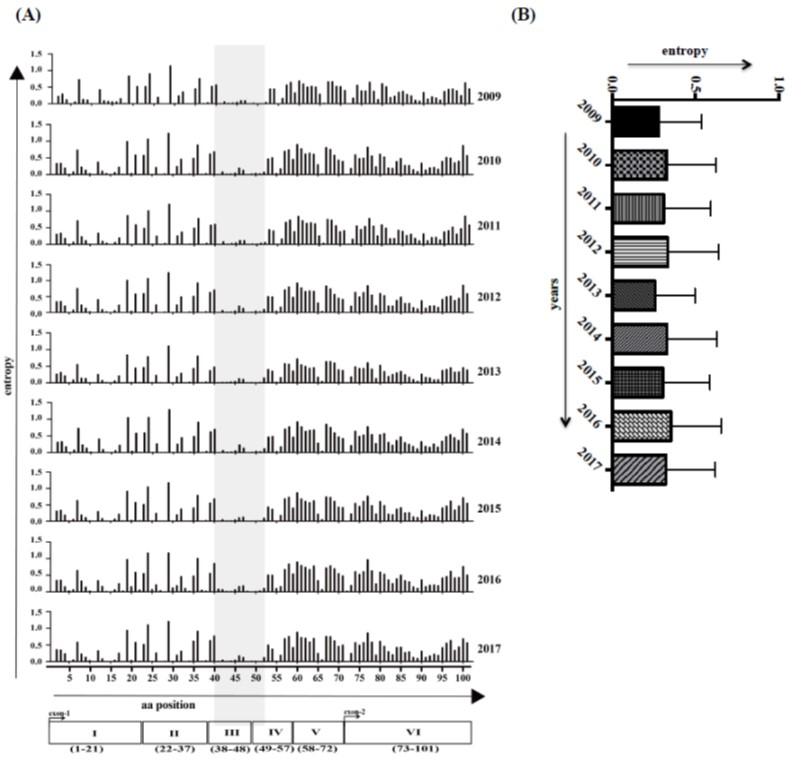
Next, we analyzed the variability of amino acid along the intact open reading frame (ORF) of Tat protein using Shannon entropy score [9], which assesses the diversity in the population in a cross-sectional sense. Amino acid variations are not evenly distributed, and the average entropy score reached 0.31 over the year (Figure 2A-B), confirming that Tat protein has not as variable as other proteins such as Env and Vpu [9, 16]. Less variability was observed in domain I-IV, while higher in domain V-VI, an indication of the functional importance among the domains. Remarkably, position 38 to 51 (domain III and a partial region of domain IV) show relatively conserved (shaded area on Figure 2A) and these regions of Tat are known to play a role for functional internalization into the host cells [17]. Sequence alignment of various HIV-1 subtype with consensus sequence from LANL database (HIV-1, M group) also revealed that Tat proteins are relatively conserved among the subtypes but contain subtype-specific signature amino acids, for example at position 57, Serine (S) for type C, while Arginine (R) for type B. Interestingly, substantial variation was observed at position 29, over 2009-2017 years, but this position also has signature amino acid for different subtypes, Lysine (K) for type A, B, D, H, Cpx; Histidine (H) for type C; Arginine (R) for type F, BC; Methionine (M) for type G; and Isoleucine (I) for type AE and AG (Figure 3). Certainly, distributions of signature amino acids were more prominent in the C-terminal part of Tat than N-terminal, which is good agreement with our entropy analysis in Figure 2.
Immune cells targeted epitopes across Tat protein
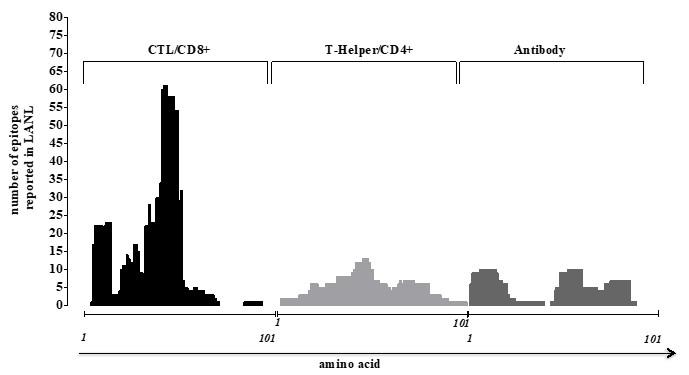
To see how human CTL/CD8+, T-helper/CD4+ and Antibody/B- cells shape HIV-1 Tat protein, we aim to search literature for reported epitopes from HIV molecular immunology database of LANL. The number of reported epitopes on Tat protein is higher by CD8+ rather than CD4+ and B-cells (Figure 4), suggesting that variability of Tat driven by CD8+ cells across the globe regardless of their subtype. We have also noticed the highly variable C-terminal region has less reported epitope by CTLs/CD8+ cells than its relatively conserved N-terminal, but the scenarios are not the same for CD4+ and B-cells.

DISCUSSION
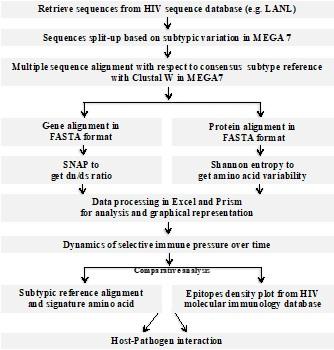
Human immunodeficiency virus type-1 (HIV-1) displays extraordinary genetic diversity, which has been a major setback in the development of vaccine and antiretroviral drugs. During host-pathogen interaction, the virus escapes immune pressures while creating a mutation (SNP) or shed certain regions of HIV-1 (i.e. glycosylation), because of its importance for functional and structural conservation to replicate [3, 18, 19]. Improvements in DNA sequencing technologies, variety of statistical tools and the availability of large sequence datasets of HIV-infected individuals (LANL), allow us to employ population-based genetic association in academia using HIV database tools (https://www.hiv.lanl.gov/content/sequence/HIV/HIVTools.html). In addition, the undertaken entire bioinformatics steps in this current study are summarized in Figure 5.
We have provided evidence that Tat protein under positive immune pressure by calculating dn/ds ratio (>1.5) using a global sequence database. Although, imposed differential immune pressure exists among the HIV-1 subtype, such as dn/ds ratio for subtype B is 1.71 whereas C is 1.49 over the year 2009 to 2017 (Figure 1A and 1C). The amino acid variations are prominent in the C-terminal of Tat protein (Figure 2A), even though the average entropy is 0.31, suggesting this protein is less variable than other proteins of HIV-1, for example Env, Gag, Pol, and Nef [1]. Remarkably, a region of position 38 to 51, known to play an important role of HIV-1 internalization in host cells, relatively conserved, could be an intriguing target for vaccination. Moreover, certain SNPs or signature amino acids were observed among the subtype in various positions of Tat, dominantly in the C-terminal end (Figure 3). The host immune profile may be one of the confounding factors of these signature amino acids or SNPs [3]. Epitope density plot analysis along with the Tat protein by T- and B-cells, indicate that CTLs/CD8+ cells play a major role in Tat sequence variation, which, however, needs further clarification using viral killing assay.
Hence, the development of a vaccine against HIV-1 infection represents the only realistic way to control the global expansion of the HIV-1 pandemic, especially in the developing world. Therefore, this study aimed at finding a delicate point of HIV-1 by quantifying selection pressures, identification of genetic loci undergoing adaptation/single nucleotide polymorphism (SNP) for subtype-specific landscape, finding the conserved region of gene in vivo [3, 19]. Indeed, targeting the signature SNP means targeting a very weak point of HIV-1 and this information would be useful for vaccine design focusing on HIV-1 subtype variation in the context of global perspective. Tat protein is a key player in HIV-1 infection since the virus lacking Tat has no infectivity as documented by several studies [20-22]. Interestingly, vaccination with Tat antigen in rodents and monkeys (systemic and mucosal administration) has also shown that it can activate specific humoral and cellular (including CTLs) immune responses [23, 24]. Therefore, finding SNPs and/or immunogenic conserved regions may be one of the contemporary approaches to target HIV-1, to boost the current vaccine efficacy.
CONCLUSIONS
The HIV sequence and immunology databases from Los Alamos National Laboratory is the repository of information about autologous sequences, alignments, epitopes and antibody binding sites along with associated tools for students and researchers studying HIV. Noteworthy, these datasets were generated and published by various laboratories around the world from HIV-1 infected human patients. Therefore, in this current study we have shown here, how to use these information’s from this open resource (e.g. LANL) to draw a prediction map/dynamics (Figure 5) of immune pressure on HIV-1, as a model pathogen.
ACKNOWLEDGEMENT
We like to thank SUST Research Centre for their funding and other technical support all through this study. We also like to thank administrative and technical staffs in Dept. of Biochemistry and Molecular biology, SUST, to set up computer and Internet facilities to complete the sequence analysis. We like to special thanks to Tanvir Hossain, Lecturer, for his kind support in the computer lab and valuable suggestions, Dept. of Biochemistry and Molecular biology, SUST, Sylhet, Bangladesh.
AUTHOR CONTRIBUTIONS
ZH, MH and AA were involved in conception and design of the experiments. MH, AA, RM, MAN and ZSN contributed to perform the sequence analysis. ZH, MH and AA analyzed data. ZH wrote the manuscript and MH and AA help in revising the manuscript. MAH and MWM contributed to revising it critically for important intellectual content. ZH made the final approval of the version to be published.
CONFLICTS OF INTEREST
Authors declared that they have no conflict of interest.
References
- [1]Korber-Irrgang B. HIV signature and sequence variation analysis. computational analysis of HIV molecular sequences. Allen G Rodrigo and Gerald H Learn, eds Dordrecht, Netherlands: Kluwer Academic Publishers. 2000;Chapter 4:55-72.
- [2]Mascola JR, Haynes BF. HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunological reviews. 2013;254:225-44.
- [3]Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PloS one. 2009;4:e6687.
- [4]Jost S, Altfeld M. Evasion from NK cell-mediated immune responses by HIV-1. Microbes and infection. 2012;14:904-15.
- [5]Mujib S, Liu J, Rahman A, Schwartz JA, Bonner P, Yue FY, et al. Comprehensive Cross-Clade Characterization of Antibody-Mediated Recognition, Complement-Mediated Lysis, and Cell-Mediated Cytotoxicity of HIV-1 Envelope-Specific Antibodies toward Eradication of the HIV-1 Reservoir. Journal of virology. 2017;91.
- [6]Zheng N, Fujiwara M, Ueno T, Oka S, Takiguchi M. Strong ability of Nef-specific CD4+ cytotoxic T cells to suppress human immunodeficiency virus type 1 (HIV-1) replication in HIV-1-infected CD4+ T cells and macrophages. Journal of virology. 2009;83:7668-77.
- [7]Cafaro A, Tripiciano A, Picconi O, Sgadari C, Moretti S, Butto S, et al. Anti-Tat Immunity in HIV-1 Infection: Effects of Naturally Occurring and Vaccine-Induced Antibodies Against Tat on the Course of the Disease. Vaccines. 2019;7.
- [8]Brady J, Kashanchi F. Tat gets the “green” light on transcription initiation. Retrovirology. 2005;2:69.
- [9]Yusim K, Kesmir C, Gaschen B, Addo MM, Altfeld M, Brunak S, et al. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. Journal of virology. 2002;76:8757-68.
- [10]Li L, Dahiya S, Kortagere S, Aiamkitsumrit B, Cunningham D, Pirrone V, et al. Impact of Tat Genetic Variation on HIV-1 Disease. Advances in virology. 2012;2012:123605.
- [11]Kurosu T, Mukai T, Komoto S, Ibrahim MS, Li YG, Kobayashi T, et al. Human immunodeficiency virus type 1 subtype C exhibits higher transactivation activity of Tat than subtypes B and E. Microbiology and immunology. 2002;46:787-99.
- [12]Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution. 2016;33:1870-4.
- [13]Wrabl JO, Grishin NV. Gaps in structurally similar proteins: towards improvement of multiple sequence alignment. Proteins. 2004;54:71-87.
- [14]Ronsard L, Lata S, Singh J, Ramachandran VG, Das S, Banerjea AC. Molecular and genetic characterization of natural HIV-1 Tat Exon-1 variants from North India and their functional implications. PloS one. 2014;9:e85452.
- [15]Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. Aids. 2006;20:W13-23.
- [16]Hasan Z, Carlson JM, Gatanaga H, Le AQ, Brumme CJ, Oka S, et al. Minor contribution of HLA class I-associated selective pressure to the variability of HIV-1 accessory protein Vpu. Biochemical and biophysical research communications. 2012;421:291-5.
- [17]Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends in cell biology. 2000;10:290-5.
- [18]Silver ZA, Antonopoulos A, Haslam SM, Dell A, Dickinson GM, Seaman MS, et al. Discovery of O-Linked Carbohydrate on HIV-1 Envelope and Its Role in Shielding against One Category of Broadly Neutralizing Antibodies. Cell reports. 2020;30:1862-9 e4.
- [19]Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439-43.
- [20]Zhou M, Deng L, Kashanchi F, Brady JN, Shatkin AJ, Kumar A. The Tat/TAR-dependent phosphorylation of RNA polymerase II C-terminal domain stimulates cotranscriptional capping of HIV-1 mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12666-71.
- [21]Meltzer B, Dabbagh D, Guo J, Kashanchi F, Tyagi M, Wu Y. Tat controls transcriptional persistence of unintegrated HIV genome in primary human macrophages. Virology. 2018;518:241-52.
- [22]Nicoli F, Gallerani E, Sforza F, Finessi V, Chachage M, Geldmacher C, et al. The HIV-1 Tat protein affects human CD4+ T-cell programing and activation, and favors the differentiation of naive CD4+ T cells. Aids. 2018;32:575-81.
- [23]Tikhonov I, Ruckwardt TJ, Hatfield GS, Pauza CD. Tat-neutralizing antibodies in vaccinated macaques. Journal of virology. 2003;77:3157-66.
- [24]Gavioli R, Cellini S, Castaldello A, Voltan R, Gallerani E, Gagliardoni F, et al. The Tat protein broadens T cell responses directed to the HIV-1 antigens Gag and Env: implications for the design of new vaccination strategies against AIDS. Vaccine. 2008;26:727-37.