Insilico analysis of γ- aminobutyric acid transaminase (GABA-T) of Brassica napus (Rape)
Abstract
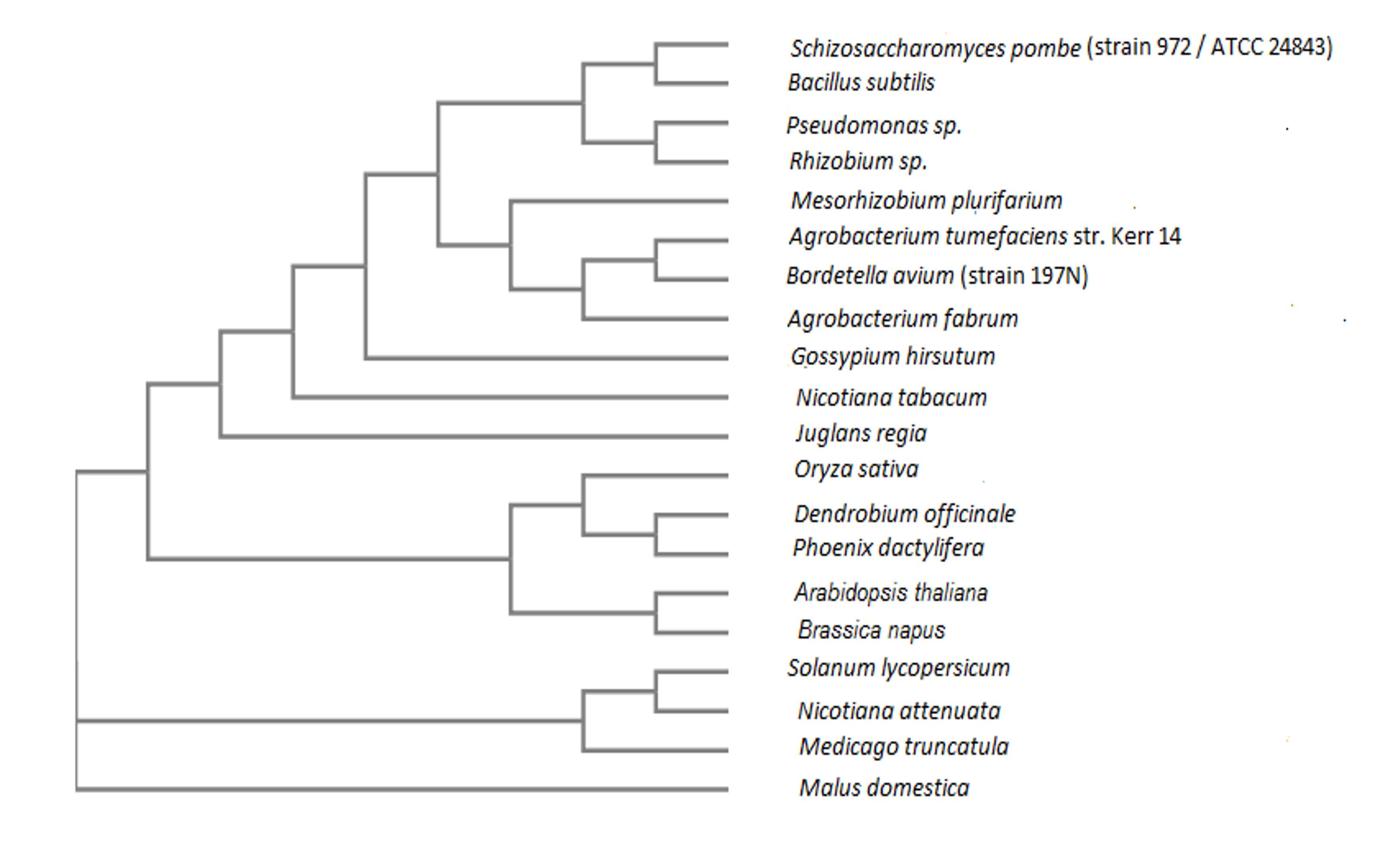
γ- aminobutyric acid (GABA) is an essential metabolite which plays a crucial role in signal transmission, stress metabolism, and some other activities also reported. Although the actual function of GABA shunt is not clearly understood. Three key enzymes, gamma aminobutyric acid transaminase (GABA-T), succinic semialdehyde dehydrogenase and succinic semialdehyde reductase are involved in GABA shunt mechanism en route from glutamate to the tricarboxylic acid cycle (TCA)which could pave the way of GABA shunt action. The enzyme gamma aminobutyric acid transaminase (GABA-T) could also play a key role in GABA shunt action by converting GABA to succinic semialdehyde (SSA).In this study, the protein sequence of γ-aminobutyric acid transaminase of Brassica napus (Rape) was retrieved from UniProt protein database and analyzed GABA-T enzyme using different bioinformatics tools and servers to analyze the physiochemical properties, amino acid composition, conformational states, and 3D structure. We found that our experimental protein sequence was very unstable, and the graph of Local Quality Estimate showed that the sequence was porn to mutation and value of Z score was above two in comparison with a non-redundant set of PDB structure. In addition, the phylogenetic tree revealed that GABA-T of Dendrobium officinale, Phoenix dactylifera, Oryza sativa, Arabidopsis thaliana and Brassica napus evolved from a common ancestor gene.
INTRODUCTION
γ-aminobutyric acid (GABA) is a ubiquitous, non-protein amino acid involved in the metabolism of stress and transmission of signal in plants [1]. It is found in both unicellular and multicellular organisms and is involved in many aspects of plant life cycle [2]. It first discovered in plants over half a century ago [3]. This molecule has been intensively investigated in mammals in which it acts as a neurotransmitter in the central nervous system [4]. Besides neurotransmission, it may work in carbon: nitrogen metabolism and responding during stress. However, much less is known about the role of GABA and its transport across the plasma membrane in plants [5]. In plants, the enzyme gamma aminobutyric acid transaminase (GABA-T) involves in catalyzing for the conversion of GABA to succinic semialdehyde (SSA) [6]. The catabolism occurs in the mitochondrial matrix of multi-cellular organisms by the action of GABA transaminase (GABA-T; EC 2.6.1.19) [2]. Two branched pathways may catabolize this succinic semialdehyde (SSA): first case, it uses SSA dehydrogenase (SSADH; EC 1.2.1.16) to form succinate and enters into tricarboxylic acid cycle, and second case, utilizing SSA reductase to form α-hydroxybutyric acid [7]. In this study, we focus on the activity of gamma aminobutyric acid transaminase. The result from Renault et al. (2013) showed that GABA-T deficiency during salt stress causes root and hypocotyl developmental defects and alterations of cell wall composition of Arabidopsis [4]. (GABA-T) GABA is a metabolite en route from glutamate to the TCA cycle, which provides succinate and NADH to the respiratory machinery [8]. The way from glutamate to succinate is known as the GABA shunt [3]. The activity of GABA shunt is induced by both abiotic and biotic stress [8]. The GABA and the GABA shunt necessary for regulation of cytosolic pH, nitrogen storage and metabolism, protection against oxidative stress, development, and deterrence of insects [7, 9, 10]. Interest in plant GABA increased mainly following observations of rapid elevation of its levels under abiotic stresses. Nevertheless, the roles of GABA under these conditions are not clear [3]. It is postulated that due to the presence of stimuli or abiotic stress, accumulation of GABA is increased which enables in attachment of cell surface binding site that generate transient Ca2+ increase and transport into cells via high affinity GABA transporters (e.g., GAT1;) [5], which may activate glutamic acid decarboxylase enzyme via Ca2+/ calmodulin complex [11]. GABA rapidly accumulates under various stress conditions such as low temperature, mechanical stimulation, and oxygen deficiency [9]. In this research, the insilico analysis of the gamma-aminobutyric acid transaminase (GABA-T) may be helpful in understanding the molecular mechanism of underlying the γ-aminobutyric acid (GABA) action or any future genetic manipulation for their target application.
MATERIALS AND METHODS
Data retrieval
The protein sequences were retrieved from the web server UniProt (Universal Protein Resource, a database of protein sequence and functional information) which is a freely accessible database of protein sequence. Our target protein sequence was gamma-aminobutyric acid transaminase (GABA-T) of Brassica napus (Rape), a crucial enzyme of GABA shunt. The sequence was retrieved as FASTA file using accession code number A0A0H4AKW3. The protein sequence of Gamma-aminobutyric acid transaminase of Malus domestica (UniPort accession code J9XGP8), Solanum lycopersicum (UniPort accession code Q84P54), Oryza sativa (UniPort accession code (Q6ZH29) and Arabidopsis thaliana (UniPort accession code Q94CE5) and another 16 GABA-T sequences retrieved to construct phylogenetic tree using UniPort. The T-coffee [12] (Tree based Consistency Objective Function for alignment Evaluation) multiple sequence alignment tool was used to generate phylogenetic tree.
Prediction of primary structure
The analyses of primary structure and physiochemical properties were performed using ProtParam [13] tool from ExPasy (Expert Protein analysis system). Using the ProtParam tool, which allows the computation of various physical and chemical parameters for a given protein stored in Swiss-Prot or TrEMBL or for a user entered protein sequence. The total number of amino acids, molecular weight and atomic composition, total number of atoms, total number of negatively charged residues and total number of positively charged residues, extinction coefficients, estimated half-life, aliphatic index, instability index and grand average of hydropathicity (GRAVY) is computed.
Prediction of secondary structure
The secondary structure of a protein is predicted using SOPMA [14] (Self- Optimized Prediction Method with Alignment) tool from PRABI Rhone-Alpes Bioinformatics Center (https://npsa-prabi.ibcp.fr/cgi bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). This tool evaluates the percentage of alpha helices, extended strand, beta turn and random coils. It uses homology methodology. According to percentage secondary structure is predicted. Number of conformational states can be given as either 4 (helix, sheet, turn, coil) or as 3 (helix, sheet, coil) [15]. The SOPMA shows two graphs, the first graph of SOPMA result anticipates the prediction and the second graph consist of outcome curves for all of the predicted states.
3D structure prediction
The tertiary structure of the protein sequence was analyzed by SWISS MODELING [16], is a structural bioinformatics web-server dedicated to homology modeling of protein 3D structures. The SWISS MODEL is very conservative but reliable. The reliability of a model depends on the availability of highly matched template and some other factors. The selection of a template relies on several parameters like sequence identity, oligomeric state, GMQE (Global Quality Model Estimate), ligand and QSQE (Quaternary Structure Prediction). It allows FASTA, Clustal, plain string, or a valid UniProtKB AC. SWISS-MODEL expert system features are automated modelling of homo-oligomeric assemblies, modeling of essential metal ions and biologically relevant ligands in protein structures, and model reliability estimates based on the QMEAN local score function. Global quality estimates based torsion angle, solvation potentiality, all atom angles, Cβ deviations, and QMEAN value. After building our model, we download as PDB format. This PDB data file subjected for PROCHECK analysis in PDBsum [17]. From PDBsum, we analyzed and validated our designed model based on different parameter like Ramachandran plot statistics, ligand, and protein-protein interaction. RAMPAGE [18] tool was used to compare PROCHECK data of Ramachandran plot.
RESULTS AND DISCUSSION
Primary structure and phylogenetic analysis
Our target protein sequence was 498 amino acids long, where highest amount of alanine (9.4%) and leucine (9.0%), and lowest amount of tryptophan (1.2%), and cysteine (1.2%) were presented (Table 1). We found negatively charged residues for (Asp + Glu) and positively charged residues for (Arg + Lys) where total number of atoms was 7750 including structural formula C2482H3879N651O719S19. Extinction coefficients were in units of M-1 cm-1 at 280 nm measured in water. The stability index was 50.4 which indicated that the protein was unstable (Table 2).
In the phylogenetic tree (Figure 1), we found that the GABA-T of Brassica napusis was closely related with Arabidopsis thaliana where GABA-T of Dendrobium officinale was closely related with Phoenix dactylifera. The phylogenetic tree revealed that GABA-T of Oryza sativa Dendrobium officinale, Phoenix dactylifera, Arabidopsis thaliana and Brassica napus were evolved from a common ancestor gene.
Table 1. Amino acid composition of GABA-T obtained from ProtPram [13].
Table 2. The different parameters of a primary structure obtained from ProtPram [13].
Secondary structure prediction
The secondary structure of protein was predicted using SOPMA (Self- Optimized Prediction Method with Alignment) tool [19]. This tool evaluated the percentage of alpha helices (43.17%), extended strand (13.65%), beta turn (7.43%) and random coils (35.74%). The windows output width, similarity threshold and number of conformational state were 17, 8 and 4, respectively. From PDBsum, we found the numbers of interface residues were 70 in chain A, and 68 in chain B, in addition, number of salt bridge, number of hydrogen bonds and number of non-bonded contracts were 2, 28 and 424, respectively. The topology of the secondary structure is presented in Figure 2, designed by PDBsum.
3D structure prediction
Our target protein sequence found total 1041 templates for homology modeling. To reveal the high sequence similarity, 50 templates were displayed (Table 3) on the screen from SWISS-MODEL Template Library (SMTL) [20]. The first templates (5ghg.1.A), showed the highest sequence identity (50.12) which was aminotransferase class-III and used for building target homology modeling. This template was found by Basic Local Alignment Search Tool (BLAST), for high Sequence similarity 0.44. The global quality estimation of the resulting model, (GMQE) is 0.71, the value of QSQE of the template, is 0.92, and the value of QMEAN is -1.67. The 3D structure of GABA-T showed in figure 2 including ligand.
In Figure 3. A is the target 3D structure of our protein developed by PDBsum, where chain A (green colored), chain B (red colored) and yellow colored in the center is the ligand (PMP, 4′-Deoxy-4′-aminopyridoxal-5′-Phosphate, PMP) where figure B. and C showed the Ramachandran Plot before PROCHECK. This plot favored 93.4%, which reflects an acceptable homology modeling of target protein. After PROCHECK, the Ramachandran Plot favored 85.9% in figure D and Table 4.
Table 3. The 50 templates from SWISS-MODEL Template Library (SMTL)
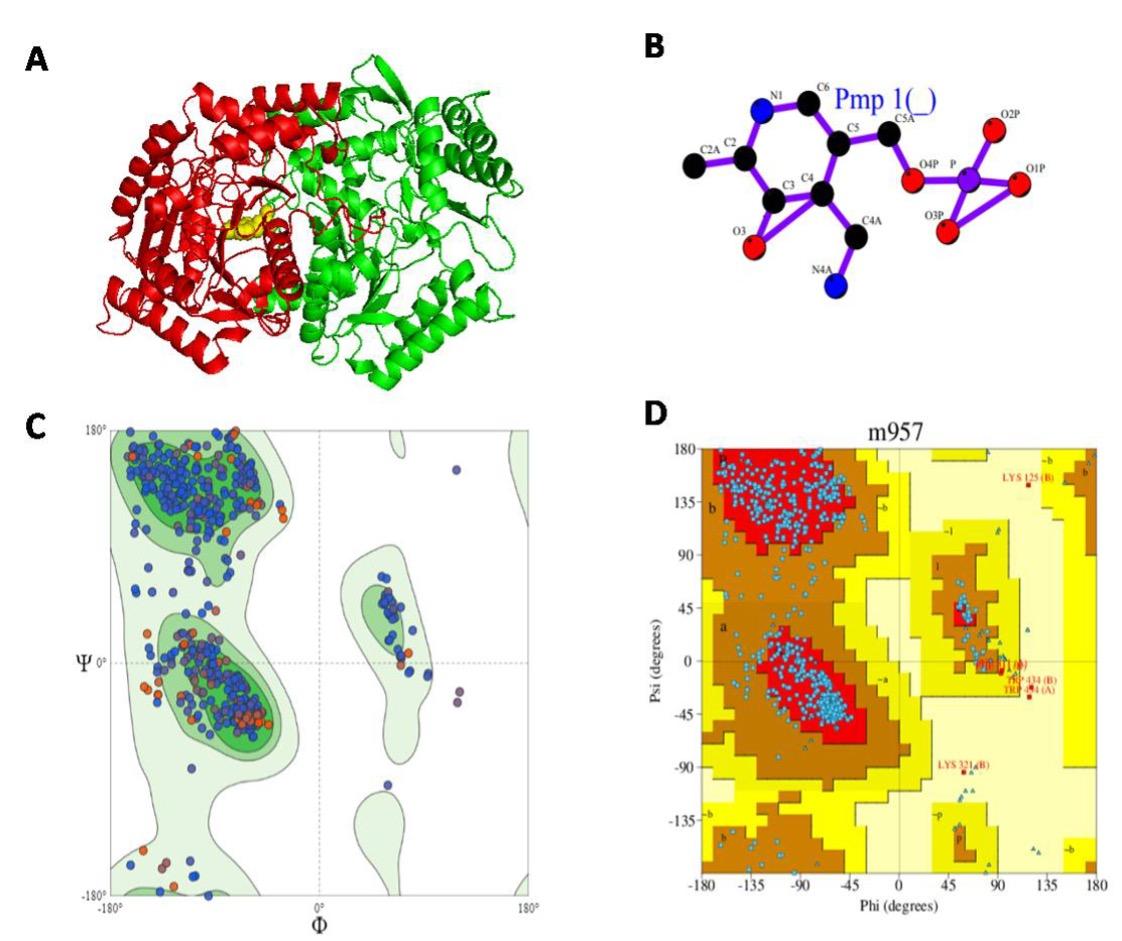
Table 4. PROCHECK analyses of Ramachandran Plot [19].
Ramachandran plot analyses
In Ramachandran Plot PROCHECK analyses, we found total 854 residues 854 including Glycine ( 66), Proline (53), End-residues (excl. Glycine and Proline) (4) and other highest number of residues were found in non-glycine and non-proline residues. The most favored region in Ramachandran plot was 85.9% (Table 3). It suggested that the percentage of most favored region (above 90%) could be desirable for good modeling.
Molprobity statistics
In MolProbity statistics, we found that the Ramachandran plot favoured 93.4% and the residues with bad angles were 0.0055 which was below 0.1% and values of bad bonds were 0.00029 (near to zero) (Table 5). But the C-Beta deviations were quite higher which might be zero or near to zero. The value of Ramachandran Outliers and Rotamer Outliers were very satisfactory.
Table 5. MolProbity results from Swiss Homology modeling using MolProbity in Phenix version 1.13. [20].
Assessment of the Ramachandran Plot by Rampage
In RAMPAGE, we found number of residues in favored region 94.1%, (~98.0% expected) which was quite higher from Swiss Modeling and the number of residues in allowed region was 5.5%, (~2.0% expected). However, in PROCHECK analyses, we found 13.6% of residues in allowed region [Combined of additional allowed regions (a,b,l,p) and generously allowed regions (~a,~b,~l,~p)], and number of residues in outlier region, (0.4%).
CONCLUSION
γ-aminobutyric acid (GABA) is not only a metabolite which plays a significant role in signal transmission, stress metabolism, regulation of cytosolic pH etc. The activity of GABA extensively studied in mammals but in plants the role of GABA is less known. This insilico analysis of the key enzyme gamma aminobutyric acid transaminase (GABA-T) could be helpful in understanding the role of GABA shunt. This protein sequence was unstable and porn to mutate and phylogenetic tree revealed that GABA-Tof Malus domestica, Solanum lycopersicum, Oryza sativa, Arabidopsis thaliana and Brassica napus were evolved from a common ancestor gene.
CONFLICT OF INTEREST
The author declares that no conflict of interest exists.
References
- [1]Clark SM, Di Leo R, Van Cauwenberghe OR, Mullen RT, Shelp BJ. Subcellular localization and expression of multiple tomato γ-aminobutyrate transaminases that utilize both pyruvate and glyoxylate. J Exp Bot. 2009; 60(11): 3255–3267.
- [2]Michaeli S, Fromm H. Closing the loop on the GABA shunt in plants: are GABA metabolism and signaling entwined? Front Plant Sci. 2015; 6: 419.
- [3]Steward FC, Thompson JF, and Dent CE. γ-aminobutyricacid: a constituent of the potato tuber? Science 1949; 110: 439–440.
- [4]Renault H, El Amrani A, Berger A, Mouille G, Soubigou-Taconnat L, Bouchereau A, Deleu C.γ-Aminobutyric acid transaminase deficiency impairs central carbon metabolism and leads to cell wall defects during salt stress in Arabidopsis. Plant Cell Environ 2013; 36, 1009–1018.
- [5]Meyer A, Eskandari S, Grallath S, and Rentsch D. At GAT1, a high affinity transporter for γ-aminobutyricacidin Arabidopsisthaliana. J.Biol. Chem. 2006; 281: 7197–7204.
- [6]Shimajiri Y, Ozaki K, Kainou K, Akama K. Differential subcellular localization, enzymatic properties and expression patterns of γ-aminobutyric acid transaminases (GABA-Ts) in rice (Oryza sativa). J Plant Physiol.2013; 170 (2): 196-201.
- [7]Shelp BJ, Bown AW, and McLean MD. Metabolism and functions of gamma-aminobutyric acid. Trends Plant Sci. 1999; 4(11): 446-452.
- [8]Bouché N, Fait A, Bouchez D, Møller SG, Fromm H. Mitochondrial succinic-semialdehyde dehydrogenase of the γ-aminobutyrate shunt is required to restrict level of reactive oxygen intermediate in plants. Proc Natl Acad Sci U S A. 2003; 100. 6843-8.
- [9]Bouché N, Fromm H. GABA in plants: Just a metabolite? Trends plant sci. 2004; 9: 110-5.
- [10]Bouche N, Fait A, Bouchez D, Moller, SG, and Fromm, H. Natl. Acad. Sci. U. S. A.2003:100, 6843–6848.
- [11]Baum G, Chen Y, Arazi T, Takatsuji H, and Fromm H. A plant glutamate decarboxylase containing a calmodulin binding domain. Cloning, sequence, and function alanalysis. J. Biol. Chem. 1993; 268: 19610–19617.
- [12]Magis C, Taly JF, Bussotti G, Chang JM, Di Tommaso P, Erb I, Espinosa-Carrasco J, Notredame C.T-Coffee: Tree-based consistency objective function for alignment evaluation. Methods Mol Biol.2014; 1079: 117-29.
- [13]Elisabeth G, Christine H, Alexandre G, S’everine D, Marc RW, Ron DA, Amos B. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook pp 571-60.
- [14]Geourjon C, Deléage G SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci.1995; 11:681–684.
- [15]Singh N, Upadhyay S, Jaiswar A, Mishra N. In silico Analysis of Protein. J Bioinform, Genomics, Proteomics.2016: 1(2): 1007.
- [16]Schwede T, Kopp J, Guex N, Peitsch MC. “SWISS-MODEL: an automated protein homology-modeling server”. Nucleic Acids Res. 2003; 31: 3381–3385..
- [17]Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. “PDBsum: a Web-based database of summaries and analyses of all PDB structures”. Trends Biochem Sci. 1997; 22(12): 488–90.
- [18]Lovell SC1, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins: Structure, Function & Genetics. 2002; 50: 437-450.
- [19]Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. 2011. Bioinformatics. 2011; 27(3): 343-50.
- [20]Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer, TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. 2018. Nucleic Acids Res. 46(W1): W296-W303.